 2021, Vol. 32
2021, Vol. 32 


2 Faculty of Chemical Engineering, Kunming University of Science and Technology, Kunming 650500, China
Pollutants, such as sulfur dioxide (SO2), nitric oxides (NOx), trace heavy metals and particulate matters (PMs) are typical components in industrial exhaust flue gas, which are harmful to both the environment and human health. SO2 and NOx are the main precursors of acid rain and toxic chemical smog [1, 2]. Elemental mercury (Hg0) is among the most toxic heavy metals owing to high toxicity, volatility and bio-accumulation [3]. The flue gas contains three mercury species: Hg0, oxidized mercury (Hg2+), and particle bound mercury (Hgp) [4]. Most of the Hgp and Hg2+ can be effectively collected by conventional air-pollution control devices (APCDs) such as electrostatic precipitators, fabric filters, and wet flue-gas desulfurization (WFGD) equipment, which are broadly installed in plants around the world [5]. However, Hg0 is relatively stable in the atmosphere, and (due to its chemical inertness, water insolubility, and volatility) is hardly removed by existing APCDs [6].
The current techniques for removing SO2, NOx, and Hg0 are selective catalytic reduction (SCR) [7, 8], WFGD [9], and activated-carbon injection [10, 11]. However, individual pollutant-treatment systems are expensive to construct and operate, complex, prone to instability, and require a high occupational area [12]. The current technologies for simultaneous removal of SO2, NOx and Hg0 include carbon-based material adsorption [13-15], photocatalytic oxidation [16, 17], wet scrubbing [18-20], NH3-SCR [7, 8], and non-thermal plasma (NTP) technologies [21, 22]. Carbon-based material adsorption and wet scrubbing have the disadvantages of secondary pollution and high investment costs. Photocatalytic oxidation and NTP have the disadvantage of limited industrial applications. Compared with these technologies, NH3-SCR catalytic technology has the advantages of high efficiency, low energy consumption and low operating cost. This makes the catalytic method to remove SO2, NOx and Hg0 has become the main research direction for air pollution control. Initially, this research focused on SO2, NOx, and Hg0 removal by catalysts without carriers, but the removal effect was non-ideal [23]. In later studies, the SO2, NOx, and Hg0 removal was improved by loading active components (transition metals, rare metals, precious metals) on various carriers (carbon materials, metal oxides, metal–oxide frameworks (MOFs), zeolites) [24-28]. This review concludes the advantages and disadvantages of simultaneous removal methods, introduces the catalysts prepared by conventional and new carriers, and discusses the low-temperature performances of the catalysts. The mechanism of pollutant removal by catalysts is also discussed, as well as the possible research directions and future prospects.
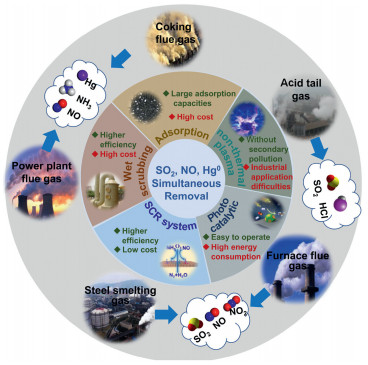
2. Removal technologiesSimultaneous removal technologies include carbon-based material adsorption [13-15], photocatalytic oxidation [16, 17], wet scrubbing [18-20], NH3-SCR [7, 8], and non-thermal plasma (NTP) technologies [21, 22]. The removal techniques are summarized in Fig. 1. In carbon-based material adsorption, pollutants are often removed by modified activated carbon, active coke, or activated-carbon fibers, which have porous structures and strong adsorption capacities. However, carbon-based material adsorption is limited by high operating cost and quality degradation of fly ash [29]. Under UV/visible radiation, pollutants are oxidized by positively charged holes on the photocatalyst surface. A photocatalytic oxidation system can be worked at relatively low temperature and pressure [30], but the UV light source raises the energy consumption. In wet scrubbing technology, the oxidant solution in the scrubbing system (such as NaClO2, KMnO4/NaOH or H2O2) can oxidize NO and Hg0 to soluble NO2 and Hg2+, which can then be removed by chemical solutions. However, this method produces large volumes of wastewater and high secondary pollution. NTP is a recently developed gas-phase oxidation method that removes multi-pollutants from flue gas [31, 32]. NTP technologies remove pollutants quickly and efficiently, operating at atmosphere pressure and room temperature with no secondary pollution [33, 34]. Their defects are high energy consumption and limited amount of flue gas; moreover, the high-voltage electricity poses a safety hazard that limits its industrial application [35]. Synergistic removal of NO and Hg0 by existing NH3-SCR systems promises to overcome the disadvantages of previous simultaneous multi-pollutant-removal technologies. In the NH3-SCR system, NO is reduced to N2 and H2O by NH3, and Hg0 is oxidized to Hg2+ by O2. The generated Hg2+ attaches to PM surfaces and is finally removed by electrostatic precipitators (ESP) or wet flue-gas desulfurization (WFGD) [36]. The NH3-SCR system improves the purification efficiency and lowers the operating costs of industrial exhaust flue-gas purification, without generating secondary pollution [12, 37, 38]. The present article systematically reviews the catalyst types, the key factors controlling the reaction processes, and the proposed reaction mechanisms of NH3-SCR technologies.

|
Download:
|
| Fig. 1. Industrial exhaust gases source and simultaneous removal of SO2, NO and Hg0 technology. | |
The existing catalysts for simultaneous removal of NO and Hg0 have been studied in the absence [23, 39, 40] and presence of carriers. Carriers can be divided into two main categories: conventional and new. Carbon-based materials [15, 41-43, 44], metal oxides [26, 27, 45-47] and silica [48, 49] are among the conventional carriers, whereas molecular sieves [50-53], MOFs [28, 54] and pillared interlayered clays (PILCs) [24, 55] are new carriers. This paper reviews the differences between the conventional and new supported catalysts in the simultaneous removal of SO2, NO and Hg. Most supported metal–oxide catalysts are prepared by the impregnation method. In the presence of different carriers, the same active component can exhibit very different activities. However, as most catalysts are studied only in the laboratory, more research on their practical applications is needed.
3.1. Catalysts 3.1.1. Unsupported catalystMany catalysts used to remove NOx, SO2 and Hg0 have been reported in the literature. Among the catalysts without carriers [23, 39, 40] are the CeO2-TiO2 (CeTi) catalysts of Li et al. [39], which oxidize elemental mercury. They found that when the weight ratio of CeO2/TiO2 is 1:2, the CeTi catalyst exhibits higher Hg0 oxidation activity in 150−250 ℃, indicating that their superior performance was conferred by the high focuses of surface cerium and oxygen. Zhang et al. [23] researched TiO2-Al2O3-CeO2 materials synthesized by the sol-gel method, which simultaneously remove NO and Hg0. They found that TiAl10Ce20 nanoparticles supplemented by 10 wt% Al2O3 removed NO and Hg0 with superior efficiency (93.41% and 80.54%, respectively) in an SCR atmosphere at 300 ℃. Song et al. [56] studied the effect of Mn, Ti and Cu doping on simultaneous removal of NO and SO2 of Fe2.5M0.5O4 catalyst. The results show that the doping of the three metal oxides can significantly increase the removal rate of NO, but has no effect on the removal rate of SO2, because SO2 can be completely removed under most conditions. Sun et al. [57] prepared a series of MFe2O4 (M=Mn, Zn, Cu, Ni, Co) catalysts to remove NO and SO2 by hydrothermal method. They found that the removal rate of SO2 at room temperature can reach 100%, and the CuFe2O4 catalyst has the highest activity compared to other catalysts. Shi et al. [58] studied the natural ferrous manganese ore to simultaneously remove NO, Hg0 and toluene from flue gas. Activity evaluation experiments show that the maximum efficiencies of NO, Hg0 oxidation are 95.29%, 83.0%. The characterization results show that the oxidation activity of manganese ore is mainly provided by Mn4+ and Fe3+ in it.
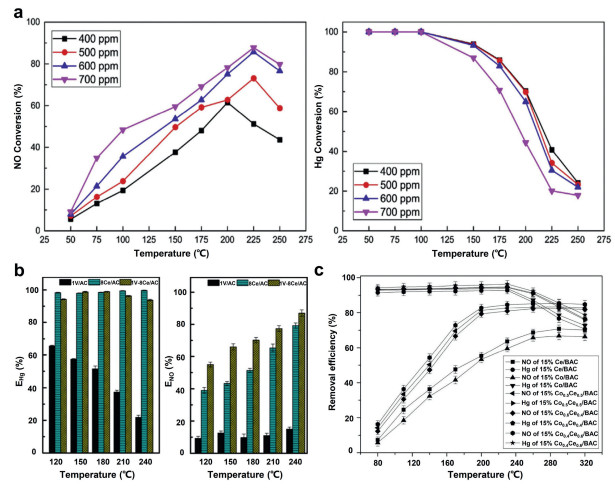
3.1.2. Catalysts based on conventional supportsCarbon-based catalysts: Carbon-based materials are the most commonly used adsorbents, which are often used to remove multi-air-pollutant from flue gas, including activated carbon, activated coke and activated carbon fiber [59-61]. Using an activated-carbon carrier, Song et al. [62] prepared 1%M-6%Y/AC (where M is Ni, Al or Mo) by equal-volume impregnation. The catalytic activity of simultaneous SO2 and NOx removal was maximized on the 1%Ni-6%Y/AC catalyst. Sumathi et al. [63] prepared a palm shell activated carbon (PSAC) adsorbent and loaded several metal oxides (Ni, V, Fe and Ce) on the ride to simultaneously remove SO2 and NOx. Among their sorbents, CeO2/PSAC yielded the maximum sorption capacity for the simultaneous removal of SO2 and NOx. Sun [64] studied the catalytic activity of copper and iron-loaded microwave coconut shell activated carbon (MCSAC) with different loadings for simultaneous denitrification and mercury removal. The 15%Cu-5%Fe/MCSAC catalyst exhibited the best removal efficiency. The NO removal rate was 86% and the mercury removal rate was 61% at 225 ℃. In addition, effect of NH3 concentration on removal of NO and Hg0 was investigated, as shown in Fig. 2a.

|
Download:
|
| Fig. 2. (a) Effect of NH3 concentration on removal of NO and Hg0 for 15%Cu–5%Fe/MCSAC catalyst. Reproduced with permission [64]. Copyright 2019, Elsevier. (b) Simultaneous removal Hg0 and NO over 1 V/AC, 8Ce/AC, and 1 V-8Ce/AC at different temperatures. Reproduced with permission [68]. Copyright 2019, American Chemical Society. (c) Effect of active ingredient on removal of NO and Hg0 for 15%Co0.4Ce0.6/BAC. Reproduced with permission [69]. Copyright 2018, Elsevier. | |
Zhang et al. [65] studied the removal effect of FeCeOx/CNTs catalyst for NO and Hg and the SO2 tolerance. They found that the NO and Hg removal rates of the catalyst were only 15.44% and 16.13%. Coating FeCeOx/CNTs catalyst with a layer of SiO2 can significantly improve the activity and SO2 tolerance of the catalyst. Li et al. [66] used activated coke as a medium to simultaneously remove SO2 and NOx from flue gas. It is found that AC still has high desulfurization and denitration efficiency after regeneration cycle. Zhao et al. [67] employed a CuO-MnOx/AC-H catalyst for the simultaneous removal of Hg0 and NO from flue gas. They pointed out the significant role of the reaction temperature in physisorption and chemical oxidation. The optimal temperature for simultaneous removal of Hg0 and NOx was 200 ℃. At this temperature, the 8%CuO-5%MnOx/AC-H catalyst achieved simultaneous removal efficiencies of 90% for mercury and 78% for NOx. Zhu et al. [68] synthesized a series of V2O5-xCeO2/AC catalysts to investigate the capabilities of the catalysts to simultaneously remove Hg0 and NO in the temperature range from 120 ℃ to 240 ℃. They discovered through experiments that compared to 1 V/AC and 1 V-8Ce/AC, 8Ce/AC catalyst has the highest Hg0 removal performance (EHg > 98%) at 120−240 ℃. The 1 V-8Ce/AC catalyst showed the best NO conversion performance compared to 1 V/AC and 8Ce/AC, 8Ce/AC, and the ENO increased gradually from 55% to 86.9% with the increase of temperature (120−240 ℃), as shown in Fig. 2b. Zhu et al. [44] used ammonium halide modified rice husk char to remove gaseous mercury. They found that the RHC adsorbent impregnated with ammonium bromide has excellent mercury removal performance, and can still reach an efficiency of about 80% for removing gaseous mercury. In addition, they found that this enhanced HgO removal adsorbent can synergistically remove SO2 and NO. Gao et al. [69] studied a series of CoOx-CeO2 loaded biomass activated carbon (CoCe/BAC) derived from maize straw. When removing both NO and Hg0 at 230 ℃, the 15%Co0.4Ce0.6/BAC sample achieved 96.8% Hg0 removal efficiency and 84.7% NO removal efficiency. Based on characterization analyses, the author attributed the best performance of 15%Co0.4Ce0.6/BAC to its largest specific surface area, lowest crystallinity and strongest redox ability than other samples in the series. The NO removal was unaffected by low SO2 concentration in the gas stream, and the NO conversion efficiency reached 80%, as shown in Fig. 2c.
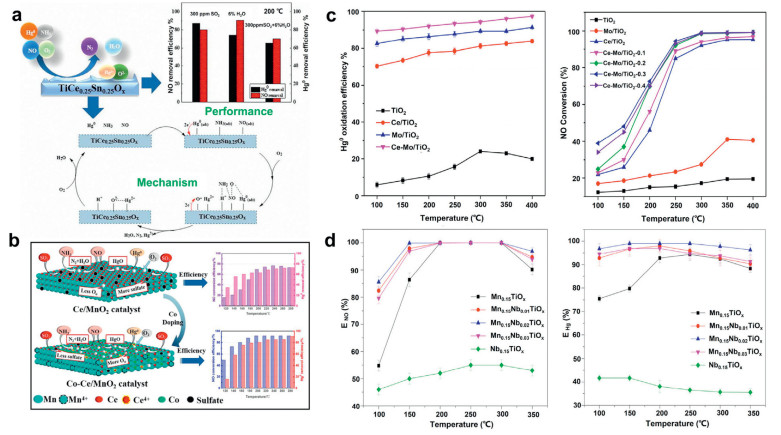
Metal-oxide-based catalysts: Titania has stable physical and chemical properties, is non-toxic, and its surface area is below 100 m2/g [70]. TiO2 has a certain acidity which enables it to be the main site of NH3 adsorption in the NH3-SCR reaction and thus beneficial to the removal of NOx. Therefore, TiO2 is a common support widely used in NH3-SCR reaction. The most prevalent commercial catalyst is V2O5-WO3/TiO2, which delivers high activity but is active only within a narrow temperature window (300–400 ℃) [71]. Zhao et al. [46] found that adding CeO2 to the V2O5-WO3/TiO2 catalyst can improve its catalytic activity for Hg0 oxidation without using HCl. The co-catalyst CeO2 shows satisfactory oxidation activity and improved water oxidation resistance. The promoted catalysts showed satisfactory oxidation activity and improved H2O resistance. Wang et al. [72] studied the oxidation catalytic performance of VWTi catalysts with different loadings of CuO. The results show that the 7%Cu/VWTi exhibited high Hg0 oxidation and a desired NO removal efficiency at 280−360 ℃. Meng et al. [73] prepared a series of Mn-V-W/TiO2 (Mn-VWT) catalysts by impregnation to simultaneously remove Hg0 and NO. The results show that Mn-VWT-400 has the best simultaneous removal performance. Its removal efficiency of Hg0 and NO reached 100% and 82%, respectively. Yang et al. [74] used CeO2 to modify V2O5/TiO2 for simultaneous removal of NO and Hg0. Experiments show that CeO2 modification not only significantly improves the SCR performance, but also significantly enhances the oxidation of Hg0. Wang et al. [75] investigated the sulfated CuCl2/TiO2 catalyst to simultaneously remove NO and Hg. By characterizing the catalyst before and after vulcanization, it was found that the CuSO4 generated on the catalyst increased the number of acid sites on the catalyst surface, thereby improving the SCR activity. CuCl2(S18)/TiO2 catalyst showed simultaneous catalytic oxidation of Hg0 (73.9%) and NO (87%) at the temperature of 325 ℃. Wang et al. [76] synthesized CeO2 supported on anatase TiO2 with high-energy facets and used it in the NH3-SCR reaction. In the activity evaluation experiment, it was found that compared with Ce/P25 catalyst, it has high SCR activity. Through the characterization, it is concluded that the unique feature of the active energy (001) facet improves the thermal stability of CeO2, and the presence of Ti3+ on the surface of TiO2 effectively promotes the development of the SCR process, both of which lead to the remarkable catalytic performance for the catalyst. Chen et al. [77] studied the SCR activity of MnCe oxide-supported titanate nanotubes (TNT). And compared with the conventional MnCe/TiO2 and V2O5/TiO2 nanoparticle catalysts, it is found that MnCe/TNTs catalysts have higher SCR activity, sulfur and water resistance. The characterization results show that the BET surface area of MnCe/TNTs is larger, and the adsorption capacity of NH3 is greatly enhanced, which is beneficial to the SCR activity. Liu et al. [78] designed the MnSmCo/Ti catalyst to simultaneously remove NO and Hg0 at low temperature. The experimental results show that the catalyst exhibits NO conversion rate of 80% and Hg0 removal rate of 100% at a gas hourly space velocity of 100, 000 h−1 in the temperature range of 150−250 ℃. Li et al. [79] prepared a series of molybdenum (Mo)-modified vanadium (V)-based selective catalytic reduction (SCR) catalyst samples using the impregnation method to remove elemental mercury (Hg0) and nitrogen oxides (NO) at the same time. Through experiments, it is found that the metal loading and the order of impregnation have no effect on the denitration activity of the catalyst, and have a particularly large effect on the mercury removal activity. The dipping sequence experiment found that MoO3(7) + WO3(3) → V2O5(0.5)/TiO2 exhibited the highest mercury removal activity. The metal loading experiment found that V2O5(0.5) + MoO3(7)/TiO2 catalyst exhibited the highest mercury removal activity. Liu et al. [80] studied the simultaneous removal of NO and Hg with Sb-modified Mn/TiO2 catalyst under low-temperature SCR conditions. The results show that the MnSb/TiO2-0.25 catalyst can achieve NO removal (> 90%) and Hg0 oxidation (> 80%) at 200−300 ℃. Li et al. [26] studied The effect of flue gas composition on the removal of Hg0 and NO by a series of CeO2 (ZrO2)/TiO2 catalysts. At 250–400 ℃, the CeO2(ZrO2)/TiO2 catalyst exhibited high Hg0 catalytic oxidation efficiency (> 95%) and high denitration efficiency (> 95%). Liu et al. [45] prepared Co/TiO2 catalysts by the sol–gel method, and determined the optimal Co loading for Hg0 catalytic oxidation (7.5%). Yang et al. [81] prepared TiCe0.25Sn0.25Ox catalyst for synergetic removal of NO and Hg0 from flue gas. Through experiments they found TiCe0.25Sn0.25Ox has excellent NO and Hg0 removal performance, and also has satisfactory SO2 tolerance, and revealed the synergistic removal mechanism and the effects of flue gas components on synergetic removal of NO and Hg0, as shown in Fig. 3a. Zhang et al. [82] loaded Co-Ce oxide on rod-like MnO2 to simultaneously remove NO and Hg. They found that the Co-Ce/MnO2 catalyst has excellent SCR activity and Hg capture efficiency. In addition, the Co-Ce/MnO2 catalyst has a high tolerance to SO2. Finally, the excellent activity and high sulfur resistance of the catalyst are explained through characterization techniques, as shown in Fig. 3b. Zhang et al. [83] researched Ce-Mo/TiO2 catalyst to simultaneously remove NO and Hg. Ce-Mo/TiO2-0.3 catalyst exhibited an excellent Hg0 oxidation and NO reduction efficiencies of over 90% at above 250 ℃. In addition, Ce-Mo/TiO2-0.3 catalyst also presented an excellent resistance to SO2 and H2O poisoning and a good stability and recyclability. The evaluation data of the catalytic activity of Ce-Mo/TiO2 is shown in Fig. 3c. Liu et al. [84] found that loading Nb on MnTiOx can significantly improve the conversion of NO and the oxidation of Hg by the catalyst. The removal efficiency of different catalysts for NO and Hg are shown in Fig. 3d. In addition to TiO2, MnO2 is also used as a carrier for simultaneous removal of NO and Hg0 from flue gas.

|
Download:
|
| Fig. 3. (a) Synergetic removal of elemental mercury and NO over TiCe0.25Sn0.25Ox catalysts. Reproduced with permission [81]. Copyright 2019, Elsevier. (b) Simultaneous removal of NO and Hg0 in flue gas over Co-Ce oxide modified rod-like MnO2 catalyst. Reproduced with permission [82]. Copyright 2020, Elsevier. (c) The Mo-Ce/TiO2 catalyst for simultaneous Hg0 oxidation and NO reduction. Reproduced with permission [83]. Copyright 2019, Elsevier. (d) Simultaneous removal of NO and Hg0 over MnaNbbTiOx catalyst. Reproduced with permission [84]. Copyright 2019, Elsevier. | |
As a stable crystalline powder, Al2O3 has good thermal stability, acid and alkali corrosion resistance, easy to form a fixed shape, and the surface area is less than 200 m2/g. These ideal properties have attracted extensive attention in the field of simultaneous removal of SO2, NOx, and Hg0. Wang et al. [47] prepared Cu and Fe-loaded Al2O3 catalysts for simultaneous removal of NO and Hg0. The characterization results show that Al2O3 as a support significantly increases the surface area and pore volume of the catalyst, improves the ability of the catalyst to adsorb gaseous reactants and therefore improves their catalytic efficiency. Moreover, Al2O3 increased the absorbed oxygen amount, indicating an increased number of active oxidative sites and a stronger oxidation ability for Hg0. Yue et al. [85] prepared a series of CuaCebZrcO3/ γ-Al2O3 catalysts by impregnation method to simultaneously remove NOx and Hg0. Their research results show that 15% Cu1.4Ce0.55Zr0.25O3/γ-Al2O3 has the highest removal efficiency of NO (93%) and Hg0 (98%).
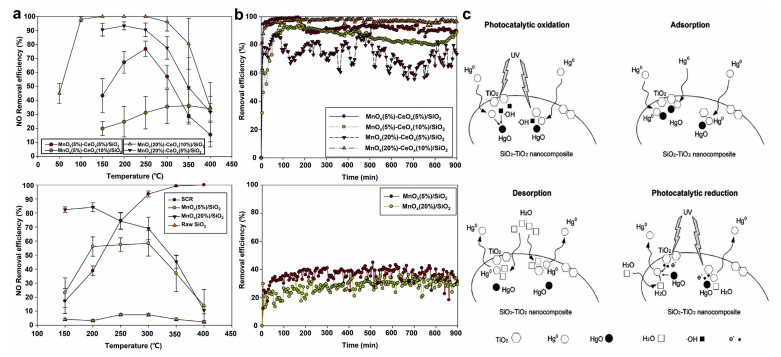
Silica based catalysts: Mesoporous silica has a large specific surface area, unique open-framework, a large pore volume and a high intrinsic surface activity, providing many adsorption sites for NO, SO2 and Hg0. Chiu et al. [86] used the impregnation method to load V, Mn and Cu oxides onto SiO2 to remove NO, SO2 and Hg. The results show that the CuOx/SiO2 catalyst exhibits the greatest Hg0 and NO removal performance. In addition, he also studied the removal of Hg0 and NO by MnOx impregnated V2O5-WO3/TiO2-SiO2 catalyst [87]. The catalysts loaded with 10% MnOx showed the best activity for the removal of Hg0 and NO. Liu et al. [49] loaded MnOx-CeOx on SiO2 to remove NO and Hg. Through the activity evaluation of different loadings of MnOx(a%)–CeOx(b%)/SiO2, it is found that 20%MnOx-10%CeOx/SiO2 has the best catalytic activity, and its mercury removal efficiency is 96% at 100−300 ℃. Secondly, they also found that the introduction of CeOx can broaden the SCR activity temperature range of MnOx/SiO2 catalysts, as shown in Figs. 4a and b. Gu et al. [88] studied the catalytic NO removal efficiency of MnFeOx/SiO2 catalysts under different Fe/Si conditions. They found that adding an appropriate amount of FeOx can significantly widen the temperature window of the catalyst. Li et al. [89] prepared a TiO2-SiO2 photocatalyst for the catalytic oxidation of Hg. Studies have shown that the catalyst has excellent catalytic oxidation activity for Hg, and the Hg removal mechanism is summarized, as shown in Fig. 4c. Chen et al. [90] used the recycled Cu modified rice-husk-derived SiO2 to remove NO and Hg. Studies have shown that 50% Cu/SiO2 has the greatest Hg removal efficiency (88.2%), and 50%Cu-10%Ce/SiO2 has 85% NO conversion at 100−300 ℃. Lin et al. [91] synthesized Cu-Mn-Ce oxide-incorporated Mesoporous Silica by hydrothermal exfoliation of silicate to remove NO and Hg. The research results show that Cu2Mn8 exhibits the highest Hg removal efficiency, and Ce doping can improve the NO removal efficiency.

|
Download:
|
| Fig. 4. (a) Removal of NO with MnOx-CeOx/SiO2. Reproduced with permission [49]. Copyright 2017, Elsevier. (b) Removal of Hg0 with MnOx-CeOx/SiO2. Reproduced with permission [49]. Copyright 2017, Elsevier. (c) Mechanisms of Hg capture and reemission on the surface of SiO2−TiO2 nanocomposite. Reproduced with permission [89]. Copyright 2006, Elsevier. | |
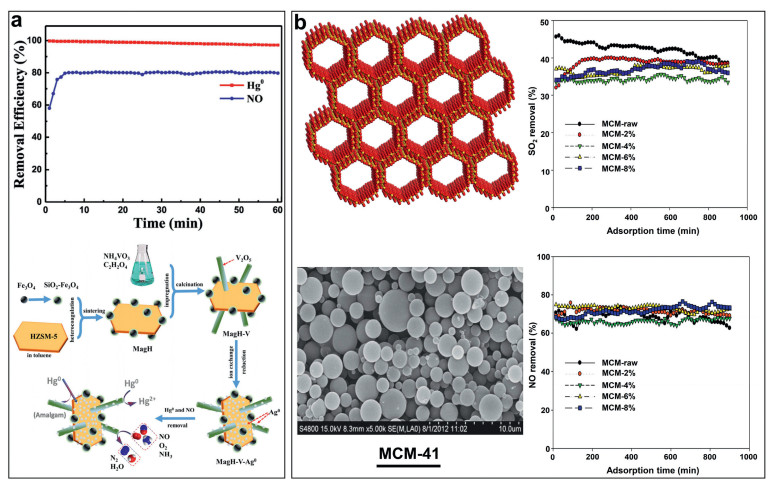
Molecular sieves-based catalysts: Zeolite-based materials have large specific surface area, good thermal stability, unique framework structure, excellent mechanical strength and adsorption, so they are considered as ideal carriers. Zeolite molecular sieves can chemically adsorb SO2, Hg and NO on the surface. The metal elements loaded on the surface of the zeolite molecular sieve and embedded in its pores can promote the oxidation or reduction of SO2, Hg and NO. Cao et al. [53] loaded Fe3O4, silver and V2O5 on HZSM-5 by dipping. The experimental results show that at 150 ℃, the removal rate of Hg by the catalyst is 97%, the removal rate of Hg0 and NO is 80%. Finally, the mechanism of simultaneous removal of NO and Hg by the catalyst is explained, as shown in Fig. 5a. Yuan et al. [92] prepared nano-iron loaded ZSM-5 and combined vaporized H2O2 and Na2S2O8, and Ca(OH)2 solution to simultaneously remove SO2, NO and Hg0. Their research results show that when the mass of the catalyst is 0.3 g, the removal rate of SO2, NO and Hg0 reaches the highest, respectively 99.9%, 96.5% and 90.1%. Fan et al. [93] prepared Cu/HZSM-5 to simultaneously remove Hg0 and NO. The experimental results show that the loading of copper significantly improves the activity of the catalyst, and the catalyst with a loading of 6% shows the best activity. Zhang et al. [67] used ZSM-5 modified with Mn-Fe to remove Hg at low temperatures and found that the introduction of Mn/Fe can effectively improve the mercury removal efficiency of ZSM-5. The catalyst with a Mn/Fe molar ratio of 1:2 has a 100% removal rate for Hg at 200−250 ℃. Applying an immersion method, Chui et al. [51] used the CuCl2/MCM prepared by the impregnation method is used for the simultaneous removal of SO2 and NO. The experimental results show that the loading of CuCl2 reduces the removal efficiency of SO2 by 4%, but increases the removal efficiency of NO by 11%, as shown in Fig. 5b.

|
Download:
|
| Fig. 5. (a) magnetically responsive catalytic sorbent and its use for removal of Hg0 and NO. Reproduced with permission [53]. Copyright 2017, Elsevier. (b) The synergistic effect of CuCl2 impregnated zeolites on simultaneous removal of Hg0, SO2 and NO. Reproduced with permission [51]. Copyright 2014, Elsevier. | |
Silico–aluminum–phosphate (SAPO) is a microporous crystal composed of three kinds of tetrahedral units, namely, PO4+, AlO4− and SiO2. Its pore structure can be controlled by changing different synthesis conditions and silicon content [94, 95]. SAPO are widely used as adsorbents, catalysts, and catalyst carriers. It is a new type of catalytic material with excellent shape selectivity, thermal stability and damp-heat stability. Because the framework is negatively charged, it has exchangeable cations and also exhibits proton acidity. SAPO can adjust the amount of acid by introducing various impurity atoms, so it can exhibit a catalytic performance ranging from medium strong acid to strong acid. It is well known that copper additives can improve the activity of SAPO-34, and copper-infused SAPO is very promising in industrial applications. Fickel et al. [94] showed that the NO conversion efficiency of Cu-infused SAPO approaches 100% at 200–400 ℃. Wang et al. [50] found that the content of Si and Al affects the content of Cu2+ species on the Cu/SAPO-34 catalyst. In addition, the content of Si can affect the content of active sites (Si or Al) and the number of acid sites of the catalyst. To affect the SCR activity of the catalyst. Pang et al. [52] prepared Mn-exchanged SAPO-34 catalysts for low-temperature SCR of NO with NH3. Among their samples, 4Mn/SAPO-34 achieved the highest SCR activity in the 120–210 ℃ temperature range. The characteristic results suggested that species related to Mn3+ and Mn4+ were the active sites of the catalyst.
MOFs-based catalysts: MOFs are intriguing porous materials with the advantage of adjustable pore size that are self-assembled by organic ligands and metal ions [96]. The adsorption performance of MOFs is boosted by sufficient porosity, large surface area, and versatile structure. In addition, MOFs promote the dispersion of active components, providing a transportation channel for catalytic reactants. The metal centers in MOFs are evenly distributed and highly dispersed, concentrating their activity in certain catalytic reactions. The advantages of MOF carriers have been exploited in the removal of SO2, NO and Hg0 from flue gas.
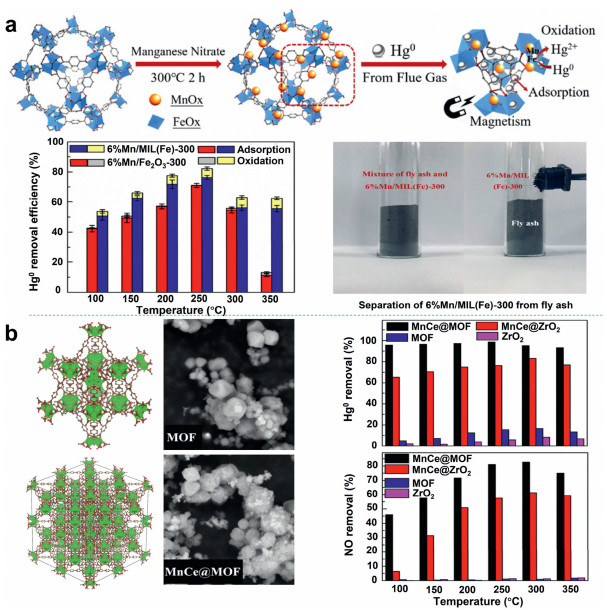
Wang et al. [97] prepared a MOF called MIL-100(Fe) and used it in the SCR reaction. Experimental results show that the NOx conversion rate of MIL-100(Fe) exceeds that of conventional V2O5-WO3/TiO2 catalysts below 300 ℃. Zhang et al. [54] investigated the MnOx-loaded MIL-100(Fe) was prepared by the impregnation method. The experimental results show that the catalyst supported by MIL-100(Fe) has higher Hg0 adsorption and oxidation capacity than the catalyst supported by Fe2O3 at all test temperatures. The Hg0 removal efficiency was 77.4% at 250 ℃. In addition, the catalyst can be easily recovered from fly ash using magnets. The catalyst activity evaluation diagram and the removal mechanism diagram are shown in Fig. 6a. Zhang et al. [28] prepared Mn-Ce supported MOF (MnCe@MOF) catalyst by impregnation method. When removing Hg0 and NO from the flue gas, the loading of Mn and Ce significantly enhanced the activity of the catalyst. Characteristic analysis shows that the MOF scaffold has a developed porous structure and a high surface area. This makes the active phase highly dispersed and thus enhances the activity of the catalyst. The sulfur and water resistance experiments show that MnCe@MOF has high resistance to SO2 and H2O. Fig. 6b shows the removal efficiency of Hg0 and NO on the MnCe@MOF catalyst and the morphology of the catalyst. The stronger adsorption of Hg0 and NH3 on MOF catalysts than on conventional materials might be explained by the special structure and Lewis acidity of MOF [98, 99].

|
Download:
|
| Fig. 6. (a) The catalyst activity evaluation diagram and the removal mechanism diagram. Reproduced with permission [54]. Copyright 2020, Elsevier. (b) The removal efficiency of Hg0 and NO on the MnCe@MOF catalyst and the morphology of the catalyst. Reproduced with permission [28]. Copyright 2017, Elsevier. | |
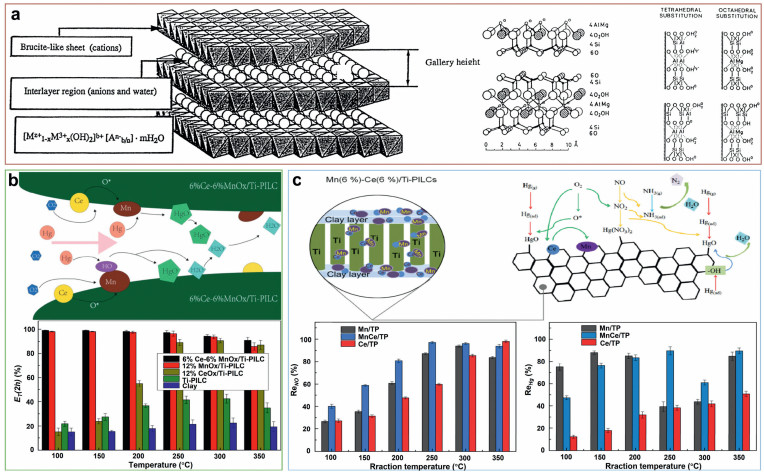
PILCs-based catalysts: PILCs is a new type of catalytic material with a two-dimensional pore-like molecular sieve structure. Schematic representation of the clay structure is shown in Fig. 7a. It has the characteristics of adjustable pore structure, high porosity, high thermal stability, hydrothermal stability and exchangeable cations, so it is widely used to make catalysts and adsorbents [55, 100-103]. Chae et al. [55] synthesized a novel SCR catalyst with a Ti–PILC support, which demonstrated higher performance than conventional catalysts. Shen et al. [24] innovatively loaded MnOx in PILCs for low-temperature NH3–SCR denitration research. At the optimal loading of MnOx/Ti–PILC (10%) and 80 ℃, the NO conversion efficiency approached 100%, with good H2O resistance. Next, Shen et al. [104] systematically studied the low-temperature denitration activity of doped Mn–MOx/Ti–PILC. They found that Ce and La additives promoted the denitrification activity [104]. He et al. [105] prepared a series of innovative Ce-Mn/Ti columnar clay (Ce-Mn/Ti-PILC) catalysts by the impregnation method for trapping elemental mercury (Hg0) at 100−350 ℃. The experimental results show that: in the temperature range of the study, the 6%Ce-6%MnOx/Ti-PILC catalyst performance is greater than 90% Hg0 removal rate. The activity evaluation data of the catalyst and the Hg removal mechanism are shown in Fig. 7b. It is found through experiments that in the first stage of the reaction, the main Hg0 capture mechanism of the catalyst is adsorption. As the reaction progresses, the oxidizing ability of Hg0 is greatly improved, which is contributed by the hydroxyl oxygen and lattice oxygen on the catalyst surface. Wang et al. [25] prepared a series of Mn-Ce/Ti-PILCs catalysts for simultaneous removal of NO and elemental mercury in simulated flue gas. The experimental results show that 6%Mn-6%Ce/Ti-PILCs exhibit excellent NO conversion rate (> 95%) and Hg0 removal efficiency (> 90%) at 250 ℃. The characterization results show that the removal activity of NO and Hg0 is strongly affected by the conversion between oxygen adsorbed on the catalyst surface and lattice oxygen. The ratio of Mn4+/Mn3+ and Ce4+/Ce3+ on the surface of the catalyst affects the conversion rate of NO and the removal of Hg0. The modified PILC behaves like a catalyst support. The NO and Hg removal efficiencies from 100 ℃ to 300 ℃ are shown in Fig. 7c.

|
Download:
|
| Fig. 7. (a) Schematic representation of the clay structure. Reproduced with permission [102]. Copyright 1998, Elsevier. (b) The removal effect of Ce-Mn/Ti-PILC on Hg and the removal mechanism diagram. Reproduced with permission [105]. Copyright 2014, American Chemical Society. (c) Simultaneous removal of NO and Hg by Ce-Mn/Ti-PILC and removal mechanism. Reproduced with permission [25]. Copyright 2015, American Chemical Society. | |
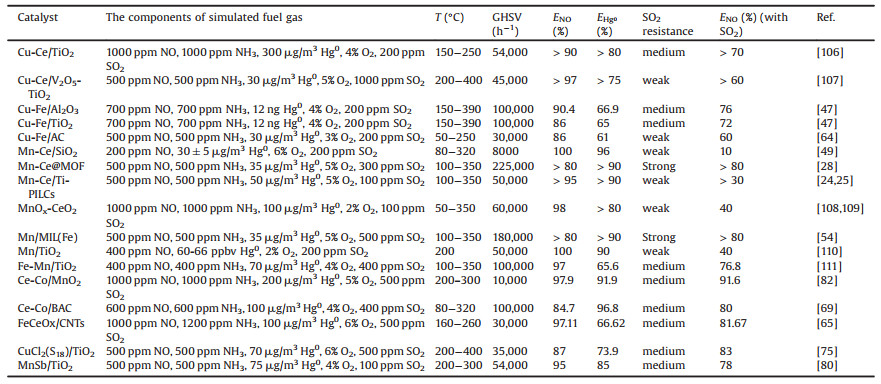
Catalyst with the same active component(s) shows very different activities in the simultaneous removal of SO2, NO, and Hg0 catalysts. Table 1 [24, 25, 28, 47, 49, 54, 64, 65, 69, 75, 80, 82, 106-111] lists the NO and Hg0 removal efficiencies and SO2 resistances of carriers with the same active components. Comparing the active metallic components among the carriers, we find that catalysts with Mn–Ce active sites are more resistant to SO2 than catalysts with other metallic active sites. Therefore, catalysts with an Mn–Ce active component are potentially valuable in industrial applications. Furthermore, Mn–Ce@MOF is more SO2-resistant than Mn–Ce/SiO2, possibly because the MOF specific surface area is higher in the former than the latter. Finally, we find that new supported catalysts are more SO2-resistant than catalysts based on conventional supports and unsupported catalyst.
|
|
Table 1 NO and Hg0 removal efficiencies and SO2 resistances of carriers with the same active components. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Real industrial exhaust gas contains species such as O2, NO, H2O, NH3, HCl, and fly ash, whose concentrations depend on the industrial type and operational conditions. Because such variable gas concentrations remarkably affect the NO reduction and Hg0 oxidation of catalysts, the impact of single flue-gas components on SO2, NO, and Hg0 removal has attracted much interest.
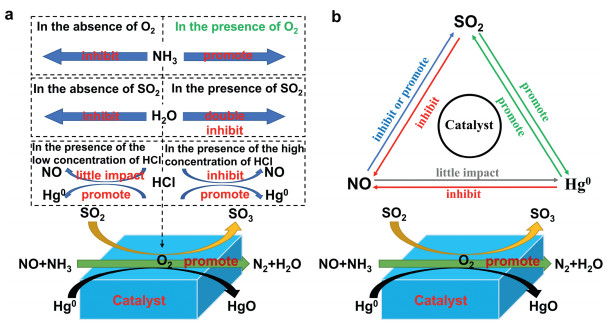
Effect of O2 : O2 is an important component in the flue gas and plays a decisive role in most catalytic reactions. [112, 113].Fig. 8a summarizes the effects of O2 on SO2, NO, and Hg0. In the absence of O2, Hg can react with the chemisorbed and lattice oxygens on the catalyst surface. As the reaction proceeds, the limited oxygen on the catalysts is gradually depleted, and the mercury removal efficiency is low. When O2 is plentiful, the Hg0 removal rate increases because the depleted lattice and chemisorbed oxygens are replaced with gas-phase O2 [114, 115]. Increasing the O2 concentration does not significantly affect the Hg0 removal efficiency. However, However, a large increase in O2 concentration will increase the rate of SO2 oxidation to SO3. The excessive generation of SO3 will not be conducive to the progress of the SCR reaction, because it reacts with NH3 and H2O to produce ammonium sulfate and viscous ammonium bisulfate, gradually occupying the active sites of the catalyst and deactivating the catalyst.

|
Download:
|
| Fig. 8. (a) A diagram showing possible effect mechanism of O2, HCl, H2O and NH3 on SO2, NO, and Hg0. (b) The interactive effects of SO2, NO, and Hg0 on the catalyst during the pollutant-removal process. | |
{kind=link}
Effect of NH3:NH3 is one of the common reducing agents in industrial applications. The NH3 content in the flue gas has a great influence on the reduction of NO and the oxidation of Hg0. Increasing the NH3/NO ratio can promote the reduction of NO in the NH3 SCR reaction, but the increase in NH3 content will cause it to compete with Hg0 for adsorption on the catalyst, thereby inhibiting the oxidation of Hg0 [26, 113, 116]. Wang et al. testified that NH3 inhibits Hg0 oxidation over a CuO-MnO2-Fe2O3/ γ-Al2O3 catalyst [117]. However, other studies have reported a significantly positive effect of NH3 on Hg0 oxidation [118]. Li et al. [119] found that NH3 consumes surface oxygen and limits the Hg0 adsorption, thereby inhibiting Hg0 oxidation over the catalyst. However, when the NH3 supply is removed and O2 is present, the inhibited Hg0 oxidation activity is completely recovered. Therefore, adding O2 at an appropriate concentration to a mixture of NO, SO2 and Hg will preserve (to some extent) the Hg0 oxidation by NH3. The mechanism by which NH3 affects the SO2, NO, and Hg0 conversion is schematized in Fig. 8a.
Effect of H2O: Water vapor is a main component in flue gases, and often leads to catalyst deactivation.Fig. 8a shows the effects of H2O on SO2, NO, and Hg0. Water vapor decreases the Hg0 oxidation and NO reduction over the supported catalyst [120], because it strongly competes with the reactive species for adsorption on the active sites of the catalyst [121]. When H2O is removed from the simulated system, the efficiencies of NO reduction and Hg0 oxidation are restored to some level slightly below the initial value.
Effect of HCl: HCl is regarded as the flue-gas component that most strongly affects the Hg0 oxidation, because the main oxidized mercury species in coal-combustion flue gas is HgCl2 [122].Fig. 8a summarizes the effect of HCl on SO2, NO, and Hg0. HCl is a critical oxidant for Hg0 oxidation, especially by metal-based catalysts [123]. Although the introduction of HCl can significantly promote the oxidation of Hg0, it cannot directly oxidize elemental mercury because it exists in a naturally reduced state [124]. Hg0 oxidation by HCl requires the assistance of oxygen. According to several studies [125, 126], In the presence of HCl, HCl and O2 will produce intermediate Cl2 and active chlorine Cl* on the catalyst, making the catalyst oxidize Hg0 through the Chlor-Deacon process. [127-129]. Wang et al. found that when present at low concentrations (10 ppm), HCl barely affects the NO conversion. However, at high concentrations, HCl prohibits the NO conversion because the chloride ions react with the metal on the catalyst surface, forming volatile metal-Cl compounds. These volatiles destroy the active sites and consequently deactivate the SCR reaction [130].
3.2.2. Interactive effect of SO2, NO, and Hg0The above-mentioned flue-gas components are mixed with various pollutants (SO2, NO, and Hg0), which might influence each other during the catalytic reaction. Therefore, to simultaneously remove multiple pollutants, many researchers have investigated the interactive effects of SO2, NO, and Hg0 during the pollutant-removal process. Their results are summarized in Fig. 8b.
Interactive effect of SO2 and NO: The effect of SO2 on NO removal in the SCR process is contentious. Some researchers [131, 132] believe that SO2 inhibits NO removal throughout the reaction system. Proponents of this view consider that SO2 reacts with metal oxides and NH3 in the presence of O2 to form metal sulfates and ammonium sulfates (such as NH4HSO4 and (NH4)2SO4). Especially, ammonium sulfates formed at low temperature on the catalyst surfaces occupy the active sites, lowering the SCR catalytic activity [133]. Other researchers [134, 135] considered that the sulfate formed by SO2 and metal oxides can enhance the acidity of the catalyst and help improve the low-temperature SCR activity of the catalyst, and some studies have found that introducing SO2 into the SCR system can increase the removal rate of NO. Thus far, the influence of NO on SO2 removal during the simultaneous removal of NO and SO2 has not been investigated [14, 136], possibly because the SO2 reaction dominates over the NH3 and O2 reactions, meaning that NO does not affect the SO2 removal.
Interactive effect of SO2 and Hg0: The effect of SO2 on Hg0 removal from the flue gas is complicated and inconclusive. Both promotional and inhibitory effects are reported in the literature [39, 137-139]. Li et al. [39] studied the effect of SO2 on the oxidation of mercury on CeO2-TiO2 catalyst. Through experiments, it is found that in the absence of oxygen in the simulated flue gas, SO2 greatly inhibits the oxidation of Hg0, but once O2 is introduced, SO2 can promote the oxidation of Hg0. Tao et al. [140] explained the anoxic experimental phenomenon as competition by Hg0 and SO2 for similar active sites on the catalyst surface; therefore, when no O2 exists in the flue gas, SO2 inhibits the Hg0 conversion. When O2 is present, the oxidation of SO2 to SO3 is mediated by the chemisorbed oxygen generated by the Ce3+-related charge imbalance on the catalyst. However, the effect of Hg on SO2 removal appears to be unreported in the literature. We speculate that Hg affects SO2 removal by the same process as SO2 affects Hg removal.
Interactive effect of NO and Hg0: Wang et al. [15] studied the effect of NO on Hg0 removal over an MnCe/TP catalyst. When the NO concentration increases from 200 ppm to 500 ppm, the removal efficiency of Hg0 will slightly decrease. Wang believed that the Hg0 oxidation was inhibited by other nonactive species such as nitrite, which might cover the active sites. Tao et al. [140] similarly found that in the absence of oxygen, increasing the NO concentration did not substantially change the Hg0 removal efficiency. When O2 was introduced to a flue gas containing NO, the Hg0 removal efficiency slightly increased and a large amount of oxidized mercury was observed in the gas flow downstream of the sample. Li et al. [39] reported the same effect of NO on Hg oxidation over a CeTi catalyst. NO alone exerted a slightly propulsive effect on Hg0 removal, especially at higher concentrations. The limited oxygen stored on the catalyst is consumed by both NO and Hg0 oxidation [141]. The propulsive phenomenon [69] might be explained by weak NO absorption to the sample surface. Some of these absorbed NO might react with the O2 on the metal–oxide catalyst surface, generating species such as NO+ and NO2 that can oxidize Hg0 [142].
Whether the Hg0 removal also affects NO removal is a pertinent question. Several researchers [25, 69, 112, 143] who studied the effect of Hg0 on NO removal reached a consensual conclusion: Hg0 exerts only a slight effect on the reduction of NO. The inhibitory effect has been attributed to two possible causes. First, Hg0 might compete with NO and NH3 for adsorption to catalytic sites [69]. Second, the oxidation reaction of Hg0 might accumulate HgO on the sample surface. If the accumulated Hg0 covers some active sites, the NO removal efficiency will be slightly reduced [23, 106, 112, 144].
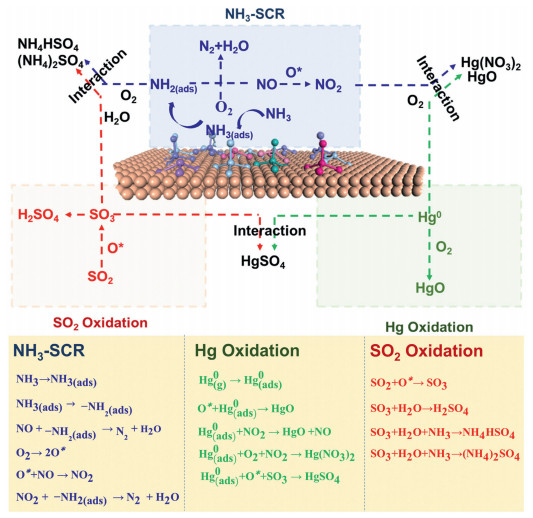
3.3. Removal mechanismBecause the supported catalyst carrier generally has a rich specific surface area and a good pore structure, the supported active component is evenly dispersed on the surface and through the pores of the carrier. A supported catalyst ensures high catalytic activity for flue-gas removal. Thus far, no complete SO2, NO, and Hg0 removal mechanism has been proposed. The mechanism of removal of SO2, NO and Hg0 under the catalyst action is summarized in Fig. 9. The pathways of SO2, NO and Hg0 removal are demonstrated in Fig. 9 with equations.

|
Download:
|
| Fig. 9. The mechanism of synergistic removal of SO2, NO and Hg0 by catalysts. (the adsorbed surface oxygen is indicated as O*). | |
{kind=link}
This paper presented the modern supported catalysts used in the removal of SO2, NO and Hg0 from flue gas. The advantages and disadvantages of the formation technologies, the effects of crucial factors, and the removal mechanisms of the supported catalysts were carefully discussed. The technologies of carbon-based material adsorption technologies, photocatalytic oxidation technologies, wet scrubbing technologies, and NTP technologies face several problems: Large energy consumption, large equipment footprint, and secondary pollution. Solid-catalytic technology, which allows higher utilization of apparatus, incurs lower operating costs, and requires less investment than multi-pollutant simultaneous removal technologies, has been widely investigated, and many supported catalysts have been researched in the past decade. MOF carriers for the low-temperature removal of NO and Hg0 provide much higher SO2 resistance than traditional supported catalysts (carbon-based materials, metal oxides, and silica). In addition, the composition of real flue gas is very complicated and remarkably affects the NO reduction and Hg0 oxidation. The removal efficiencies of NO, SO2, and Hg0 are enhanced in the presence of O2. NH3 inhibits Hg0 oxidation and NO reduction in the absence of O2, but promotes these reactions in the presence of O2. H2O inhibits Hg0 oxidation and NO reduction by competitively adsorbing to the active sites. After exploring the surface chemical processes, this review clarified the mechanisms of SO2, NO and Hg0 removal, and proposed a theory of catalytic multi-pollutant purification by new supported catalysts. Reviewing the past solid catalytic technology and the current technological development direction, we finally proposed the future research direction. At present, catalysts based on new supports have advantages in sulfur resistance and denitrification and mercury removal compared with catalysts based on conventional supports. Among the catalysts based on the new carrier, the new MOFs material as the carrier has higher sulfur resistance at low temperature than other traditional materials such as carbon materials and metal oxides. This is because its regular pore structure and large surface area make the supported metal oxides highly dispersed. Compared with other materials, its sulfur resistance is poor at high temperature. This is because the thermal stability of MOFs at high temperatures is poor. The molecular sieve material has a synergistic removal effect of thimerosal compared to other materials at high temperature. In summary, the high-temperature activity of MOFs-based supported catalysts and the low-temperature activity of molecular sieve-based supported catalysts are worthy of in-depth study in the gas purification technology for removing SO2, NOx and Hg0 from industrial exhaust gas.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (Nos. 52000093, 51968034, 41807373 and 21667015), National Key R & D Program of China (No. 2018YFC0213400), China Postdoctoral Science Foundation (Nos. 2020T130271, 2019M663911XB) and Open Fund of National Engineering Laboratory for Mobile Source Emission Control Technology (No. NELMS2019B03).
| [1] |
J.P. Zheng, China Statistical Yearbook. Beijing: China Statistics Press, 2004.
|
| [2] |
C.F. You, X.C. Xu, Energy 35 (2010) 4467-4472. DOI:10.1016/j.energy.2009.04.019 |
| [3] |
J.K. Xie, Z. Qu, N.Q. Yan, et al., J. Hazard. Mater. 261 (2013) 206-213. DOI:10.1016/j.jhazmat.2013.07.027 |
| [4] |
Y. Zhuang, J.S. Thompson, C.J. Zygarlicke, J.H. Pavlish, Environ. Sci. Technol. 38 (2004) 5803-5808. DOI:10.1021/es030683t |
| [5] |
C.L. Senior, J.J. Helble, A.F. Sarofim, FuelProcess.Technol. 65-66 (2000) 263-288. |
| [6] |
F. Scala, H.L. Clack, J. Hazard. Mater. 152 (2008) 616-623. DOI:10.1016/j.jhazmat.2007.07.024 |
| [7] |
K. Krishna, M. Makkee, Appl. Catal. B 59 (2005) 35-44. DOI:10.1016/j.apcatb.2005.01.003 |
| [8] |
Z.B. Wu, B.Q. Jiang, Y. Liu, Appl. Catal. B 79 (2008) 347-355. DOI:10.1016/j.apcatb.2007.09.039 |
| [9] |
V.K. Sharma, M. Sohn, G.A.K. Anquandah, N. Nesnas, Chemosphere 87 (2012) 644-648. DOI:10.1016/j.chemosphere.2012.01.019 |
| [10] |
H.Q. Yang, Z.H. Xu, M.H. Fan, A.E. Bland, R.R. Judkins, J. Hazard. Mater. 146 (2007) 1-11. DOI:10.1016/j.jhazmat.2007.04.113 |
| [11] |
M. Rallo, M.A. Lopez-Anton, M.L. Contreras, M.M. Maroto-Valer, Environ. Sci. Pollut. Res. 19 (2012) 1084-1096. DOI:10.1007/s11356-011-0658-2 |
| [12] |
A.P. Jones, J.W. Hoffmann, D.N. Smith, T.J. Feeley, J.T. Murphy, Environ. Sci. Technol. 41 (2007) 1365-1371. DOI:10.1021/es0617340 |
| [13] |
J.Y. Zhang, C.T. Li, L.K. Zhao, et al., Chem. Eng. J. 313 (2017) 1535-1547. DOI:10.1016/j.cej.2016.11.039 |
| [14] |
J.R. Ma, Z.Y. Liu, Q.Y. Liu, et al., Fuel Process. Technol. 89 (2008) 242-248. DOI:10.1016/j.fuproc.2007.11.003 |
| [15] |
Q.Y. Liu, Z.Y. Liu, Fuel 108 (2013) 149-158. DOI:10.1016/j.fuel.2011.05.015 |
| [16] |
Y. Yuan, J.Y. Zhang, H.L. Li, et al., Chem. Eng. J. 192 (2012) 21-28. DOI:10.1016/j.cej.2012.03.043 |
| [17] |
C.Y. Su, X. Ran, J.L. Hu, C.L. Shao, Environ. Sci. Technol. 47 (2013) 11562-11568. DOI:10.1021/es4025595 |
| [18] |
N.D. Hutson, R. Krzyzynska, R.K. Srivastava, Ind. Eng. Chem. Res. 47 (2008) 5825-5831. DOI:10.1021/ie800339p |
| [19] |
F. Ping, C.P. Cen, Z.X. Tang, et al., Chen.. Chem. Eng. J. 168 (2011) 52-59. DOI:10.1016/j.cej.2010.12.030 |
| [20] |
P. Fang, C.P. Cen, X.M. Wang, et al., Fuel Process. Technol. 106 (2013) 645-653. DOI:10.1016/j.fuproc.2012.09.060 |
| [21] |
J. Jeong, J. Jurng, Chemosphere 68 (2007) 2007-2010. DOI:10.1016/j.chemosphere.2007.01.044 |
| [22] |
Y. Qi, H.M. Yang, K.S. Zeng, Z.W. Zhang, Y. Gang, J. Environ. Sci. 11 (2007) 1393-1397. |
| [23] |
J.Y. Zhang, C.T. Li, L.K. Zhao, et al., Chem. Eng. J. 313 (2017) 1535-1547. DOI:10.1016/j.cej.2016.11.039 |
| [24] |
B.X. Shen, H.Q. Ma, Y. Yao, J. Environ. Sci. 024 (2012) 499-506. DOI:10.1016/S1001-0742(11)60756-0 |
| [25] |
Y.Y. Wang, B.X. Shen, C. He, S.J. Yue, F.M. Wang, Environ. Sci. Technol. 49 (2015) 9355-9363. DOI:10.1021/acs.est.5b01435 |
| [26] |
Z. Li, Y.S. Shen, X.H. Li, S.M. Zhu, M. Hu, Catal. Commun. 82 (2016) 55-60. DOI:10.1016/j.catcom.2016.04.019 |
| [27] |
Y.J. Yang, J. Liu, B. Zhang, et al., Chem. Eng. J. 317 (2017) 758-765. DOI:10.1016/j.cej.2017.02.060 |
| [28] |
X. Zhang, B.X. Shen, F. Shen, et al., Chem. Eng. J. 326 (2017) 551-560. DOI:10.1016/j.cej.2017.05.128 |
| [29] |
E.J. Granite, M.C. Freeman, R.A. Hargis, J.O.D. William, H.W. Pennline, J. Environ. Manage. 84 (2007) 628-634. DOI:10.1016/j.jenvman.2006.06.022 |
| [30] |
R. Thiruvenkatachari, S. Vigneswaran, I.S. Moon, Korean J. Chem. Eng. 25 (2008) 64-72. DOI:10.1007/s11814-008-0011-8 |
| [31] |
J.S. Chang, K. Urashima, Y.X. Tong, et al., J. Electrost. 57 (2003) 313-323. DOI:10.1016/S0304-3886(02)00168-7 |
| [32] |
Y. Byun, K.B. Ko, M. Cho, et al., Chemosphere 72 (2008) 652-658. DOI:10.1016/j.chemosphere.2008.02.021 |
| [33] |
H.B. Ma, P. Chen, M.L. Zhang, X.Y. Lin, R. Ruan, Plasma Chem. Plasma. Process. 22 (2002) 239-254. DOI:10.1023/A:1014895409454 |
| [34] |
W.C. Wang, Z.B. Zhao, L. Feng, W. Su, J. Electrost. 63 (2005) 155-164. DOI:10.1016/j.elstat.2004.10.002 |
| [35] |
S. Ma, Y. Zhao, J. Yang, et al., Renew. Sust. Energ. Rev. 67 (2017) 791-810. DOI:10.1016/j.rser.2016.09.066 |
| [36] |
H. Wang, B. Yuan, R.L. Hao, Y. Zhao, X.P. Wang, Chem. Eng. J. 378 (2019) 122155. DOI:10.1016/j.cej.2019.122155 |
| [37] |
A.S. Negreira, J. Wilcox, J. Phys. Chem. C 117 (2013) 24397-24406. DOI:10.1021/jp407794g |
| [38] |
G.M. Zeng, L.K. Zhao, Y.E. Xie, et al., Catal. Sci. Technol. 5 (2015) 3459-3472. DOI:10.1039/C5CY00219B |
| [39] |
H.L. Li, C.Y. Wu, Y. Li, J.Y. Zhang, Environ. Sci. Technol. 45 (2011) 7394-7400. DOI:10.1021/es2007808 |
| [40] |
D. Jampaiah, K.M. Tur, P. Venkataswamy, et al., RSC Adv. 5 (2015) 30331-30341. DOI:10.1039/C4RA16787B |
| [41] |
M.A.T. Izquierdo, B. Rubio, C. Mayoral, J.M. Andrés, Fuel 82 (2003) 147-151. DOI:10.1016/S0016-2361(02)00249-1 |
| [42] |
X.P. Fan, C.T. Li, G.M. Zeng, et al., Energy Fuels 24 (2010) 4250-4254. DOI:10.1021/ef100377f |
| [43] |
K. Jastrzab, Fuel Process. Technol. 104 (2012) 371-377. DOI:10.1016/j.fuproc.2012.06.011 |
| [44] |
C. Zhu, Y.F. Duan, C.Y. Wu, et al., Fuel 172 (2016) 160-169. DOI:10.1016/j.fuel.2015.12.061 |
| [45] |
L. Yue, Y.J. Wang, H.Q. Wang, Z.B. Wu, Catal. Commun. 12 (2011) 0-1294. |
| [46] |
L.K. Zhao, C.T. Li, J. Zhang, et al., Fuel 153 (2015) 361-369. DOI:10.1016/j.fuel.2015.03.001 |
| [47] |
D.H. Wang, Q. Yao, S. Liu, S.E. Hui, Y.N. Niu, J. Energy Inst. 92 (2018) 1852-1863. |
| [48] |
C.H. Chiu, T.H. Kuo, T.C. Chang, et al., Int. J. Coal Geol. 170 (2016) 60-68. |
| [49] |
K.H. Liu, M.Y. Chen, Y.C. Tsai, H.P. Lin, H.C. Hsi, Catal. Today 297 (2017) 104-112. DOI:10.1016/j.cattod.2017.03.037 |
| [50] |
J. Wang, T. Yu, X. Wang, et al., Appl. Catal. B 127 (2012) 137-147. DOI:10.1016/j.apcatb.2012.08.016 |
| [51] |
C.H. Chiu, H.C. Hsi, C.C. Lin, Fuel Process. Technol. 126 (2014) 138-144. DOI:10.1016/j.fuproc.2014.04.031 |
| [52] |
C.K. Pang, Y.Q. Zhuo, Q.Y. Weng, RSC Adv. 7 (2017) 32146-32154. DOI:10.1039/C7RA05165D |
| [53] |
T.T. Cao, Z.J. Zhou, Q. Chen, et al., Fuel Process. Technol. 160 (2017) 158-169. DOI:10.1016/j.fuproc.2017.02.022 |
| [54] |
X. Zhang, Q.Q. Shi, B.X. Shen, Z.Z. Hu, X.Q. Zhang, J. Hazard. Mater. 381 (2020) 121003. DOI:10.1016/j.jhazmat.2019.121003 |
| [55] |
H.J. Chae, I.S. Nam, S.W. Ham, S.B. Hong, Appl. Catal. B 53 (2004) 117-126. DOI:10.1016/j.apcatb.2004.04.018 |
| [56] |
Z.J. Song, B. Wang, W. Yang, et al., Chem. Eng. J. 386 (2020) 123883. DOI:10.1016/j.cej.2019.123883 |
| [57] |
H.R. Sun, D.X. Li, RSC Adv. 10 (2020) 25155-25164. DOI:10.1039/D0RA04392C |
| [58] |
L. Shi, Y.F. Chang, L.B. Qin, et al., New J. Chem. 43 (2019) 17486-17493. DOI:10.1039/C9NJ03697K |
| [59] |
J.M. Stencel, A.M. Rubel, Coal Sci. Technol. 24 (1995) 1791-1794. |
| [60] |
K. Tsuji, I. Shiraishi, Fuel 76 (1997) 555-560. DOI:10.1016/S0016-2361(97)00022-7 |
| [61] |
H.H. Tseng, M.Y. Wey, Y.S. Liang, K.H. Chen, Carbon 41 (2003) 1079-1085. DOI:10.1016/S0008-6223(03)00017-4 |
| [62] |
Y.J. Song, T. Wang, Y. Zhao, et al., J. Fuel Chem. Tech. 044 (2016) 1112-1118. |
| [63] |
S. Sumathi, S. Bhatia, K.T. Lee, A.R. Mohamed, J. Hazard. Mater. 176 (2010) 1093-1096. DOI:10.1016/j.jhazmat.2009.11.037 |
| [64] |
X. Sun, L.N. Sun, Y. Liu, et al., J. Energy Inst. 93 (2020) 87-98. DOI:10.1016/j.joei.2019.04.006 |
| [65] |
H.W. Zhang, M.Z. Zhang, L.F. Hao, et al., Fuel Process. Technol. 201 (2020) 106342. DOI:10.1016/j.fuproc.2020.106342 |
| [66] |
Y. Li, X. Zhang, F.L. Huang, et al., Fuel 275 (2020) 117862. DOI:10.1016/j.fuel.2020.117862 |
| [67] |
B. Zhao, H.H. Yi, X.L. Tang, et al., J. Hazard. Mater. 364 (2019) 700-709. DOI:10.1016/j.jhazmat.2018.04.001 |
| [68] |
Y. Zhu, Y. Hou, J. Wang, et al., Environ. Sci. Technol. 53 (2019) 5521-5527. DOI:10.1021/acs.est.8b07122 |
| [69] |
L. Gao, C.T. Li, J. Zhang, et al., Chem. Eng. J. 342 (2018) 339-349. DOI:10.1016/j.cej.2018.02.100 |
| [70] |
C. Guillard, B. Beaugiraud, C. Dutriez, et al., Appl. Catal. B 39 (2002) 331-342. DOI:10.1016/S0926-3373(02)00120-0 |
| [71] |
L.J. Alemany, L. Lietti, N. Ferlazzo, et al., J. Catal. 155 (1995) 117-130. DOI:10.1006/jcat.1995.1193 |
| [72] |
H. Wang, B. Wang, J. Zhou, et al., J. Environ. Manage. 239 (2019) 17-22. DOI:10.1016/j.jenvman.2019.02.118 |
| [73] |
J. Meng, Y. Duan, P. Hu, et al., Energy Fuels 33 (2019) 8896-8906. DOI:10.1021/acs.energyfuels.9b01503 |
| [74] |
Y. Yang, W.Q. Xu, J. Wang, T.Y. Zhu, Fuel 249 (2019) 178-187. DOI:10.1016/j.fuel.2019.03.103 |
| [75] |
Y. Wang, H.U. Li, S.K. Wang, et al., Fuel Process. Technol. 188 (2019) 179-189. DOI:10.1016/j.fuproc.2019.02.009 |
| [76] |
H.Q. Wang, S. Cao, Z. Fang, et al., Appl. Surf. Sci. 330 (2015) 245-252. DOI:10.1016/j.apsusc.2014.12.163 |
| [77] |
X.B. Chen, P.L. Wang, P. Fang, et al., Fuel Process. Technol. 167 (2017) 221-228. DOI:10.1016/j.fuproc.2017.07.018 |
| [78] |
L.J. Liu, S. Su, K. Xu, et al., Proc. Combust. Inst. 38 (2021) 5331-5338. DOI:10.1016/j.proci.2020.06.295 |
| [79] |
C. Li, D. Brewe, J.Y. Lee, Appl. Catal. B 270 (2020) 118854. DOI:10.1016/j.apcatb.2020.118854 |
| [80] |
J. Liu, R.T. Guo, X. Sun, et al., Mater. Chem. Phys. 232 (2019) 88-98. DOI:10.1016/j.matchemphys.2019.04.061 |
| [81] |
B. Yang, Z. Li, Q. Huang, et al., Chem. Eng. J. 360 (2018) 990-1002. |
| [82] |
M.Z. Zhang, J. Wang, Y.H. Zhang, et al., Fuel 276 (2020) 118018. DOI:10.1016/j.fuel.2020.118018 |
| [83] |
P. Zhang, W.G. Pan, R.T. Guo, et al., J. Energy Inst. 92 (2019) 1313-1328. DOI:10.1016/j.joei.2018.10.003 |
| [84] |
J. Liu, R.T. Guo, Z.Z. Guan, et al., Int. J. Hydrogen Energy 44 (2019) 835-843. DOI:10.1016/j.ijhydene.2018.11.006 |
| [85] |
H. Yue, P. Lu, W. Su, et al., Environ. Sci. Pollut. Res. 26 (2019) 13602-13618. DOI:10.1007/s11356-019-04822-x |
| [86] |
C.H. Chiu, H.C. Hsi, H.P. Lin, Catal. Today 245 (2015) 2-9. DOI:10.1016/j.cattod.2014.09.008 |
| [87] |
C.H. Chiu, H.C. Hsi, H.P. Lin, T.C. Chang, J. Hazard. Mater. 291 (2015) 1-8. DOI:10.1016/j.jhazmat.2015.02.076 |
| [88] |
S. Gu, K. Gui, D. Ren, Y. Wei, React. Kinet. Mech. Catal. 132 (2021) 187-201. DOI:10.1007/s11144-020-01890-w |
| [89] |
Y. Li, C.Y. Wu, Environ. Sci. Technol. 40 (2006) 6444-6448. DOI:10.1021/es061228a |
| [90] |
M.Y. Chen, Y.C. Tsai, C.F. Tseng, H.P. Lin, H.C. Hsi, Aerosol Air Qual. Res. 19 (2019) 2557-2567. DOI:10.4209/aaqr.2019.09.0468 |
| [91] |
C.J. Lin, C.L. Chang, C.F. Tseng, H.P. Lin, H.C. Hsi, Aerosol Air Qual. Res. 19 (2019) 1421-1438. DOI:10.4209/aaqr.2018.10.0389 |
| [92] |
B. Yuan, Y. Zhao, X.Z. Mao, Z.H. Zheng, R.L. Hao, Fuel 262 (2020) 116567. DOI:10.1016/j.fuel.2019.116567 |
| [93] |
X.P. Fan, C.T. Li, G.M. Zeng, et al., Fuel Process. Technol. 104 (2012) 325-331. DOI:10.1016/j.fuproc.2012.06.003 |
| [94] |
D.W. Fickel, E. D'Addio, J.A. Lauterbach, R.F. Lobo, Appl. Catal. B 102 (2011) 441-448. DOI:10.1016/j.apcatb.2010.12.022 |
| [95] |
L. Wang, W. Li, G.S. Qi, D. Weng, J. Catal. 289 (2012) 21-29. DOI:10.1016/j.jcat.2012.01.012 |
| [96] |
B. Liu, S. Jie, B. Li, Prog. In.Chem. 25 (2013) 36-45. |
| [97] |
W. Peng, H.M. Zhao, S. Hong, et al., RSC Adv. 4 (2014) 48912-48919. DOI:10.1039/C4RA07028C |
| [98] |
Y. Peng, K.Z. Li, J.H. Li, Appl. Catal. B 140-141 (2013) 483-492. DOI:10.1016/j.apcatb.2013.04.043 |
| [99] |
U.S.F. Arrozi, H.W. Wijaya, A. Patah, Y. Permana, Appl. Catal. A: Gen. 506 (2015) 77-84. DOI:10.1016/j.apcata.2015.08.028 |
| [100] |
R.T. Yang, J.P. Chen, E.S. Kikkinides, L.S. Cheng, J.E. Cichanowicz, Ind. Eng. Chem. Res. 31 (1992) 1440-1445. DOI:10.1021/ie00006a003 |
| [101] |
Ohtsuka Kunio, Chem. Mat. 9 (1997) 2039-2050. DOI:10.1021/cm9605227 |
| [102] |
A. Vaccari, Catal. Today 41 (1998) 53-71. DOI:10.1016/S0920-5861(98)00038-8 |
| [103] |
S. Cheng, Catal. Today 49 (1999) 303-312. DOI:10.1016/S0920-5861(98)00437-4 |
| [104] |
B.X. Shen, Y. Yan, H.Q. Ma, T. Liu, Chin. J. Catal. 32 (2011) 1803-1811. DOI:10.1016/S1872-2067(10)60269-0 |
| [105] |
C. He, B.X. Shen, J.H. Chen, J. Cai, Environ. Sci. Technol. 48 (2014) 7891-7898. DOI:10.1021/es5007719 |
| [106] |
H.L. Li, S.K. Wu, L.Q. Li, et al., Catal. Sci. Technol. 5 (2015) 5129-5138. DOI:10.1039/C5CY00794A |
| [107] |
G.L. Chi, B.X. Shen, R.R. Yu, C. He, X. Zhang, J. Hazard. Mater. 330 (2017) 83-92. DOI:10.1016/j.jhazmat.2017.02.013 |
| [108] |
H.L. Li, Y. Wang, S.K. Wang, X. Wang, J.J. Hu, Fuel 208 (2017) 576-586. DOI:10.1016/j.fuel.2017.07.061 |
| [109] |
X. Yao, K. Ma, W. Zou, et al., Chin. J. Catal. 38 (2017) 146-159. DOI:10.1016/S1872-2067(16)62572-X |
| [110] |
J. Lei, M.S. Pavani, G.S. Panagiotis, W.T. Stephen, G.P. Neville, Energy Fuels 22 (2008) 2299-2306. DOI:10.1021/ef700533q |
| [111] |
S.B. Zhang, Y.C. Zhao, J.P. Yang, J.Y. Zhang, C.G. Zheng, Chem. Eng. J. 348 (2018) 618-629. DOI:10.1016/j.cej.2018.05.037 |
| [112] |
L. Gao, C. Li, S. Li, et al., Chem. Eng. J. 371 (2019) 781-795. DOI:10.1016/j.cej.2019.04.104 |
| [113] |
L.K. Zhao, C.T. Li, X.Y. Du, et al., Appl. Surf. Sci. 437 (2018) 390-399. DOI:10.1016/j.apsusc.2017.08.165 |
| [114] |
A. Bueno-López, K. Krishna, M. Makkee, J.A. Moulijn, J. Catal. 230 (2005) 237-248. DOI:10.1016/j.jcat.2004.11.027 |
| [115] |
L.K. Zhao, C.T. Li, Y. Wang, et al., Catal. Sci. Technol. 6 (2016) 6076-6086. DOI:10.1039/C5CY01576F |
| [116] |
B.X. Shen, S.W. Zhu, X. Zhang, et al., Fuel 224 (2018) 241-249. DOI:10.1016/j.fuel.2018.03.080 |
| [117] |
P.Y. Wang, S. Su, J. Xiang, et al., Chem. Eng. J. 225 (2013) 68-75. DOI:10.1016/j.cej.2013.03.060 |
| [118] |
M. Rallo, B. Heidel, K. Brechtel, M.M. Maroto-Valer, Chem. Eng. J. 198-199 (2012) 87-94. DOI:10.1016/j.cej.2012.05.080 |
| [119] |
H.L. Li, Y.C. Wu, Y. Li, J.Y. Zhang, Appl. Catal. B 111 (2012) 381-388. |
| [120] |
P. Zhang, W.G. Pan, R.T. Guo, et al., J. Energy Inst. 92 (2018) 1313-1328. |
| [121] |
B.X. Shen, T. Liu, Z. Ning, X.Y. Yang, L.D. Deng, J. Environ. Sci. 22 (2010) 1447-1454. DOI:10.1016/S1001-0742(09)60274-6 |
| [122] |
Z. Ye, J. Laumb, R. Liggett, M. Holmes, J. Pavlish, Fuel Process. Technol. 88 (2007) 929-934. DOI:10.1016/j.fuproc.2007.03.010 |
| [123] |
A.A. Presto, E.J. Granite, Environ. Sci. Technol. 40 (2006) 5601-5609. DOI:10.1021/es060504i |
| [124] |
H.Y. Pan, R.G. Minet, S.W. Benson, T.T. Tsotsis, Ind. Eng. Chem. Res. 33 (1994) 2996-3003. DOI:10.1021/ie00036a014 |
| [125] |
H.L. Li, C.Y. Wu, Y. Li, et al., J. Hazard. Mater. 243 (2012) 117-123. DOI:10.1016/j.jhazmat.2012.10.007 |
| [126] |
D.Y. Chen, S.J. Zhao, Z. Qu, N.Q. Yan, Fuel 217 (2018) 297-305. DOI:10.1016/j.fuel.2017.12.086 |
| [127] |
J.P. Chen, M.A. Buzanowski, R.T. Yang, J.E. Cichanowicz, J. Air Waste Manage. Assoc. 40 (1990) 1403-1409. DOI:10.1080/10473289.1990.10466793 |
| [128] |
L. Lisi, G. Lasorella, S. Malloggi, G. Russo, Appl. Catal. B 50 (2004) 251-258. DOI:10.1016/j.apcatb.2004.01.007 |
| [129] |
W.C. Jin, I.S. Nam, Appl. Catal. A: Gen. 312 (2006) 165-174. DOI:10.1016/j.apcata.2006.06.044 |
| [130] |
F. Wang, B. Shen, L. Gao, J. Yang, Fuel Process. Technol. 168 (2017) 131-139. DOI:10.1016/j.fuproc.2017.08.024 |
| [131] |
R.B. Jin, Y. Liu, Z.B. Wu, H.Q. Wang, T.T. Gu, Catal. Today 153 (2010) 84-89. DOI:10.1016/j.cattod.2010.01.039 |
| [132] |
D.W. Kwon, K.H. Park, S.C. Hong, Chem. Eng. J. 284 (2016) 315-324. DOI:10.1016/j.cej.2015.08.152 |
| [133] |
H.L. Li, C.Y. Wu, Y. Li, et al., Chem. Eng. J. 219 (2013) 319-326. DOI:10.1016/j.cej.2012.12.100 |
| [134] |
M.S. Maqbool, A.K. Pullur, H.P. Ha, Appl. Catal. B 152 (2014) 28-37. |
| [135] |
W.J. Zhang, G.F. Liu, J. Jiang, et al., Chemosphere 243 (2020) 125419. DOI:10.1016/j.chemosphere.2019.125419 |
| [136] |
Y.L. Wang, Z.G. Huang, Z.Y. Liu, Q.Y. Liu, Carbon 42 (2004) 445-448. DOI:10.1016/j.carbon.2003.11.006 |
| [137] |
L.L. Dennis, D.B. Thomas, R.N. Babu, Fuel Process. Technol. 65-66 (2000) 157-165. DOI:10.1016/S0378-3820(99)00083-1 |
| [138] |
A.N. Glenn, H. Q. Yang, C.B. Robert, et al., Fuel 82 (2003) 107-116. DOI:10.1016/S0016-2361(02)00254-5 |
| [139] |
L. Jing, W.Q. Qu, W.J. Sang, C.G. Zheng, Chem. Eng. J. 184 (2012) 163-167. DOI:10.1016/j.cej.2012.01.023 |
| [140] |
S.S. Tao, C.T. Li, X.P. Fan, et al., Chem. Eng. J. 210 (2012) 547-556. DOI:10.1016/j.cej.2012.09.028 |
| [141] |
Y. Li, P.D. Murphy, C.Y. Wu, K.W. Powers, J.C.J. Bonzongo, Environ. Sci. Technol. 42 (2008) 5304-5309. DOI:10.1021/es8000272 |
| [142] |
Y.N. Shi, S. Chen, H. Sun, Y. Shu, X. Quan, Catal. Commun. 42 (2013) 10-13. DOI:10.1016/j.catcom.2013.07.036 |
| [143] |
Y.C. Zhu, Y.Q. Hou, J.W. Wang, et al., Environ. Sci. Technol. 53 (2019) 5521-5527. DOI:10.1021/acs.est.8b07122 |
| [144] |
L. Gao, C.T. Li, P. Lu, et al., Fuel 215 (2018) 30-39. DOI:10.1016/j.fuel.2017.11.008 |