 2021, Vol. 32
2021, Vol. 32 


b School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China;
c College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450000, China
In the first two decades of the 21st century, the environmental issues and energy crisis triggered by the consumption of fossil fuels have drawn the broad attention world widely. The employment of green energy by the electrocatalysis [1-5] and photocatalysis [6-12] and other catalysis [13-15] is urgently demanded. Electrocatalytic hydrogen evolution (HER) and oxygen evolution/reduction have extensively applied in various new energy affairs, such as hydrogen energy production [16-20], fuel cells [21-24] and metal-air batteries [25-28] displaying the promising potentials in superseding the conventional fossil fuels. However, the high cost of the commercial catalysts due to the massive utilization of noble metals still renders the large-scale deployment of the electrocatalytic energy transformation devices. The nature of this issue is originated from the low specific activities of the noble metals. The metals in the traditional catalysts are in the morphology of nano particles, for example, nanotubes and nanospheres. The metal atoms inside the particles are inactive and absent in the real reactions. Furthermore, the active sites of metal particles are normally on the corner or/and edge sites resulting in the dissipation of predominant noble metals. To address this bottleneck, the downsizing strategy of the metal particles has been adopted towards catalyst fabrications. Therefore, in 2011, Zhang Tao's team first outlined the theory of a single atom catalyst (SAC). Subsequently, in 2012 Georgios Kyriakou et al. found that a single indied Pd atom on the Cu surface can greatly reduce the energy barrier of hydrogen adsorption and desorption on the Cu metal surface, so that the hydrogenation of styrene and acetylene has a very high selectivity [29]. Jian Lin et al. applied the prepared SACs (Ir1/FeOx) to the water-gas conversion reaction in 2013, and the catalyst showed an order of magnitude more activity than its corresponding nano-particle catalyst [30]. The direct conversion of methane to advanced hydrocarbons is important for energy conversion. Nonetheless, the reaction conditions required to activate a strong C-H bond tend to result in excessive oxidation of the product. Guo et al. Described a high-temperature non oxidation route in 2014 to expose methane to isolated iron sites on silica catalysts [31]. Methyl radicals are generated in the gas phase and coupled to form ethylene, aromatics and hydrogen. The separation of active sites in the monatomic catalyst avoids the surface reaction between free radicals, resulting in the deposition of solid carbon. Huan Yan et al. prepared atomically dispersed palladium on graphene using atomic layer deposition in 2015. In the selective hydrogenation reaction of 1, 3-butadiene, Pd1/graphene catalyst at a mild reaction condition of around 50 ℃, the selectivity of butene at 95% conversion rate was 100% [32]. In the following years, scientists used more common transition metals (such as iron and cobalt) to prepare a series of SACs, which showed similar catalytic performance to Pt/C catalysts in the fields of oxygen reduction reaction [33-35], hydrogen production reaction [36] and oxygen evolution reaction [37, 38], and so on [39]. Advanced oxidation processes (AOPs) are advanced water treatment technologies that use soluble oxidants (H2O2, O3, HOCl, etc.) to generate highly active free radicals to remove organic pollutants. However, the large amount of energy and chemical input requirements limit the practical application of AOPS. Recently, Cui's team has pioneered a new low-cost organic wastewater treatment technology by using SAC and successfully addressed the above two challenges faced by AOPs technology [40]. The Cu atom in graphitized carbon nitride can catalyse the activation of H2O2 to generate hydroxyl radicals at pH 7.0 without energy input, and it shows strong stability in the filtration device. An electrolytic reactor was further designed to generate H2O2 in situ from air, water and renewable energy sources.
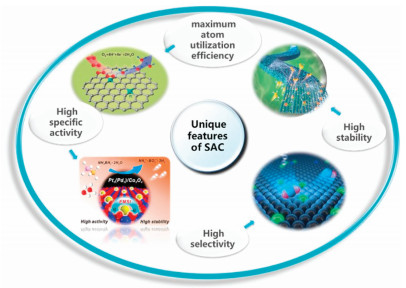
As a frontier of the catalyst, SAC has unique features [41-44] and exhibits various advantages [45]. Some recent research works have made significant progress in the evolution of greatly powerful electrocatalysts, just like reasonable regulate on their shape, size, structure, and content [24, 46-55]. In this review article, we summarized four characters of the SAC (Fig. 1).

|
Download:
|
| Fig. 1. Unique features of SAC. Reproduced with permission [41-44]. [41] Copyright 2019, American Chemical Society. [42] Copyright 2017, Elsevier. [43, 44] Copyright 2019, Wiley-VCH. | |
{kind=link}
(1) The SAC achieves the theoretical maximum atom efficiency. The single metal atom in the SAC as an active site is usually directly exposed on the support surface. In principle, on the support surface, all unsaturated coordinated sites could probably bond with specific foreign species. The theoretical atom efficiency of SAC could normally reach near 100% if we neglect the metals being embedded in the substrate, reducing the cost of SAC apparently especially for the noble metals. Jing Liu et al. announced a carbon-supported defect-anchored Pt SAC which presents high O2 reduction reaction (ORR) performance and a superhigh Pt application of 0.09 gPt/kW in fuel cells [44]. Experimental and theoretical calculations show the main active site observed for an efficient ORR process is a single Pt atom attached by four carbon atoms in a carbon divacancies. This active center has super high catalytic 4e- ORR capability. This work will facilitate the research and preparation of low-cost, pure carbon-supported, single-atom Pt-based electrocatalysts with high-efficiency catalytic oxidation reduction capabilities.
(2) The SAC displays a high specific activity in the specific reaction. Compared with atoms on supported nano-catalysts, SACs exhibit higher specific activity in most reactions. For example, Yunteng Qu et al. successfully prepared single Pt atom catalyst (Pt SAs/DG) by thermal emission strategy [56]. The Pt SAs/DG showed better HER activity than its corresponding nano-catalysts (Pt NPs/DG), which was mainly reflected in that the overpotential of Pt SAs/DG at 10 mA/cm2 was 23 mV less than that of Pt NPs/DG (38 mV). Qi Wang et al. prepared Ni based catalysts by a two-step synthesis method (first loading MoS2 nanosheets on carbon cloth, then loading Ni on MoS2 nanosheets) [36]. In HER performance test, Ni based SACs showed higher HER activity than its corresponding cluster catalyst in both acidic and alkaline environment. This high specific activity is determined by the following factors: (ⅰ) The metal atom as the active center in the SACs has a low coordination number. That the coordination number is lower than the surface atom of the nano-catalyst is more beneficial to the adsorption and activation of the reaction substrate. Therefore, the structural difference of metal atoms acting as active sites is the key aspect for the high specific activity of catalysts. Xiaoqian Wang et al. induced efficient CO2 electroreduction by regulating the coordination number of Co atomic sites [57]. The results illustrated that the Co SACs with double coordination with nitrogen atom showed the best catalytic activity, the Faraday efficiency of CO production reached 94%, and the current density was 18.1 mA/cm2 at the overpotential of 520 mV. Combined with theoretical calculations, they confirmed that lower coordination number can promote the electroreduction of CO2. (ⅱ) Metal-support interaction. In SACs, all active center metal atoms form coordination bonds with adjacent atoms on the support (usually nitrogen, oxygen and carbon). Therefore, the charge transfer between the support and the active metal atom may alter the electronic state and further tune the adsorption property of the substrate. In general, the supported nano-catalysts only exhibit such changes at the heterogenous interface, while SACs exhibit more comprehensive changes. This difference in electronic states is a profound boost to the high specific activity of monatomic catalysts. In consideration of SACs, the coordination configuration of the single atom will significantly impress their electronic structure, then regulate their catalytic activity. Recently, Yao et al. reported edge-rich Fe-N4 active sites in defective carbon for ORR [58]. Both DFT calculations and experimental results illustrate that edge-nitrogen-modified divacancies (e-ND-Fe) trapped atomic Fe motifs shows better ORR activities than intact center model. The difference is mainly coming from the local electronic redistribution and bandgap shrinkage for e-ND-Fe. Zhang et al. successfully synthesized atomically distributed Fe-N-C catalyst, and it displayed high activity as well as remarkable recyclability for the selective oxidation of the C-H bond [59]. In the atomic dispersed Fe-N-C catalyst, there are three different coordination configurations of Fe atoms, namely FeN x (x = 4–6). In light of the results, although the corresponding concentration of FeⅢN5 in the catalyst is much lower than the relative concentration of FeⅢN6 structure, FeⅢN5 with medium-spin shows the highest conversion frequency (6455 h-1). By using electronic metal-support interactions (EMSIs) to regulate the d orbital energy level of single metal atom, Junling Lu et al. designed a superior activity and stability single-atom catalyst [41]. Specifically, the activity of the catalyst shows 68 times larger than that of borane dehydrogenation for room temperature hydrogen generation those on other supports, which is mainly due to the modification of the 5d state of the single atom Pt1 on Co3O4 by the powerful EMSIs. (ⅲ) SACs present the capricious active sites from traditional catalysts in some reactions. Specifically, the support of a SAC may change the path of the catalytic reaction or the coordination atoms around the metal atom may be converted to the active sites. For example, Wang et al. considered the repercussion of transition-metal carbides (TMCs) as substrates on CO2 reduction reaction (CO2RR) on supported Pd catalysts [60]. It was proved by DFT that the TMC support changed the CO2RR performance of Pd coating by adjusting *HOCO adsorption, resulting in enhanced CO2RR activity.
(3) The SAC exhibits the high reaction pathway selectivity. Since the central metal atoms with different coordination structures present the different free energy in the elemental reactions, the reaction pathway can be manipulated by the controllable synthesis of the specific coordination structure of the atomic metals. For clean energy applications and environmental protection, it is increasingly important to reduce carbon dioxide in chemicals or fuels by using clean electricity. The challenge comes from fierce competition for hydrogen release reactions in aqueous solutions, especially for those earth-rich transition metals such as Ni, which can greatly reduce the selectivity for CO2 reduction. Separating the transition-metal single atom into the matrix of graphene can moderately adjust its catalytic behavior. It is profitable to the conversion of CO2 to CO. Haotian Wang et al. reported a catalyst using transition-metal Ni atoms as coordination center in graphene shells [42]. The catalyst reduced CO2 aqueous to CO at a high current of up to ~60 mA/mg, and the faraday efficiency was as high as 90%. The 3D atom probe was used to characterize the transition metal Ni as a single atom distributed on the graphene defect. Combining theoretical calculations, it was shown that atomic Ni exhibited a different electronic structure than metallic Ni, which promoted the transition from CO2 to CO and significantly suppressed the competitive catalytic hydrogen evolution reaction (HER). On the basis of previous Ni-graphene work, the team of Haotian Wang further researched the electrocatalytic reduction of CO2. A elementary way for synthesis single Ni atom catalysts with low cost and high yield was reported [61]. The shortcomings of traditional liquid-phase catalysis were overcome. Combining with the design of anion membrane electrode reaction device, a high-selective large-current catalytic reduction of CO2 to CO was achieved. The current density generated was higher than 100 mA/cm2, the selectivity to CO was close to 100%, and the side reaction of hydrogen evolution was about 1%. The group of Hyunjoo Lee successfully prepared a single Pt atom loaded on TiN NPs, verified that the two-electron path could selectively and efficiently produce H2O2 [62].
(4) The SAC presents high stability. The strong covalent bonds betwixt the metal atoms and support are capable to immobilize the single metal atom, preventing it from further aggregations even under the condition of high temperature or large oxidation potential. Lu Zhao et al. reported a common cascade anchoring approach for the mass production of a set of M-NC SACs with a metal content of more than 12.1 wt% without any aggregations. The excellent durability of Fe-NC SAC in alkaline media was also confirmed by the polarization curves recorded after 5000 cycles [63]. This could be ascribed to the profoundly robust coordination interactions between iron atom and the support. Contrary to Fe-NC SAC, the commercial Pt/C catalyst revealed a half-wave potential loss of 20 mV and a significant reduction in the ultimate current density after accelerated durability test (ADT). The severe dissolution/aggregation of Pt nanoparticles during ADT led to this significant reduction in Pt/C catalyst activity. Sintering resistance is also an important embodiment of good stability of SACs. Sintering leads to a reduction in the active surface area of the catalyst, and it has been proved that sintering has an adverse effect on the activity of various vital reactions, like methane oxidation, selective hydrogenation of nitroaromatic hydrocarbons, etc. [64-66]. The key to prevent sintering is to adjust the bonding strength between individual metal atoms and oxide support. Nolan J. O'Connor et al. studied the interaction between single metal atom and oxide support by statistical learning and density functional theory [67]. The results show that the adsorption energy of metal on the surface of oxide has a linear trend with the enthalpy of formation of metal oxide, and the slope of different surfaces has a linear correlation with the surface oxygen vacancy formation energy. The reducibility of each support reflects its strong adsorption capacity for metal atoms. For example, TbO2 (111) which is the most easily reducible surface has stronger adsorption capacity for metal atoms than MgO (100) which is the most difficult to reduce. In addition, in the reducible surface, the binding of metal atoms with low oxygen affinity to the support is still very weak, which leads to the low stability of SACs. These characteristics can be used to empirically screen the strength of metal support interaction, then helping to devise anti-sintering SACs. Recently, Hongying Zhuo et al. summarized the stability of single metal atom on graphene support [68]. They pointed out that the original graphene is not suitable for loading metal atoms, because the calculation results show that metal atoms are inclined to drift and form clusters in the course of the reaction. However, for the defect graphene supported metal atom, the existence of free valence of uncoordinated carbon atom in the defect can upgrade the stability of metal atom. In addition, the defect structure acts momentous part in avoiding the diffusion and migration of adsorbed metal atoms, which also has a great guiding significance for the design of catalysts. Deserve to be mentioned, the binding energy of metal atoms on the single vacancy graphene is much larger than that on the original graphene. Finally, they discussed strategies for modifying graphene substrates with non-metallic elements to enhance the catalyst stability, counting non-metallic doped original graphene, non-metallic doped defective graphene, and graphene oxide (GO). Compared with the original graphene, B/N doping can improve the binding energy of single metal atom, but the stabilizing effect on metal single atom is far less than the above defects. In the case of doped defective graphene, the boundary C atom at the vacancy position is replaced by non-metallic dopant. The stability of single metal atom on this support depends on the type of defect (single vacancy or double vacancy) and the number/type of doped nonmetallic atoms. In the case of GO, the abundant oxygen species on the surface provide anchoring sites for the active metal atoms, which prevent the migration and aggregation of single metal atoms due to strong ion and covalent interactions. In addition, it has been proved that stable O-M-O bridging bonds are usually formed in stable go supported metal atom configurations. Starting from the observation of the interesting phenomenon that platinum atoms migrate as gaseous species a high temperatures, John Jones et al. proposed to use the crystal face of cerium oxide with a specific morphology to capture single atoms, so as to construct a supported catalyst system to overcome the limitation of catalyst thermal stability of traditional catalyst [69]. In this study, based on the experimental phenomenon of the gas transfer of platinum at high temperature, the author discovered the trapping effect of cerium oxide and its different morphologies on platinum atoms, and obtained SACs with certain thermal stability. This study provides important research ideas for us to understand the formation process of SA and how to improve the thermal stability of SACs. However, whether this atom trapping mechanism is suitable for all types of SACs needs further research. Hong Wang et al. described an extensible approach to establish an electrochemically active, layered porous carbon film involving atomically dispersed semi-metallic selenium (Se) (called SeNCM) [43]. The separated Se atom is maintained by a carbon atom in the mode of a six-atom ring structure, where the Se atom is placed at the edge of graphitic. This structure is dissimilar with the formerly noted transition/noble metal SACs. SeNCM possess several advantages of positively charged selenium, an enlarged graphite layer, and the vigorous electrochemical properties. These advantages made the catalyst show higher catalytic activity than Pt/C. The catalyst further express continuing operational stability for the hydrazine oxidation reaction in actual hydrazine fuel cells. The excellent properties of SACs have attracted much attention. Therefore, the main target of this article is to give a rundown of the recent development of SAC, including the classification, preparation, characterization, application in electrocatalysts. Eventually, the current challenges and the outlooks of the SAC research will be proposed.
2. The category and preparation of SACs 2.1. The category of SACIn the past decade, SAC has developed rapidly. According to the different types of supports, the SACs could fall into the following four categories [70-74] (as shown in Fig. 2).

|
Download:
|
| Fig. 2. The HAADF–STEM image and schematic illustration of (a) crown-jewel-structured Au/Pd nanocluster catalysts. Reproduced with permission [70]. Copyright 2012, Nature Publishing Group. (b) Pt–MoS2. Reproduced with permission [71]. Copyright 2015, Royal Society of Chemistry. (c) Fe@C2N catalyst. Reproduced with permission [72]. Copyright 2018, Elsevier BV. (d) Ni SAs/N-C. Reproduced with permission [73]. Copyright 2017, American Chemical Society. (e) Synthetic route of Rh@S-1 catalyst. (f) The STEM image of Rh@S-1-H viewed along the direction [011] and the schematic diagram of the model on the same projection. Reproduced with permission [74]. Copyright 2019, Wiley-VCH. | |
{kind=link}
The SAC assembling the atomic metals on the metal support achieves the advantages of the simple preparation, low cost, and generally good catalytic performances for ORR and CO oxidation reaction. Unfortunately, since the metallic support is dissoluble in the acidic media, the application of SA@M is limited to the alkaline and neutral environment. As shown in Fig. 2a the structure with top Au atoms decorating Pd nanoclusters (Red and yellow represent gold and palladium atoms, respectively) is named as the crown-jewel-structure [70]. The HAADF-STEM image in Fig. 2a obviously illustrates the presence of columnar atoms in CJ-Au/ Pd NC with a diameter of about 2 nm. The atoms at the center and the left corners (site 1 and site 2, green circles in the image) display the ordered arrangement indicating their sluggish activity. However, the nano particle in Fig. 2a does not form an ordered atomic alignment around the lower right corners (site 3 and site 4), which is ascribed to the size mismatch between the Au and Pd atoms because of the interatomic distance between Au and Pd is 0.292 and 0.276 nm, respectively. In addition, obvious vacancies resulting from the displacement reaction are also detected in the right corners (site 3-site 6) marked by the red circles. These vacancies and unordered atomic alignments give believable proof that the substitution reaction of Au atoms does occur at the top of the Pd147NC. This CJ-structure catalyst is subsequently predicted to exhibit an unexpected glucose oxidation catalytic activity for by the DFT calculations.
2.1.2. Single metal atom anchored on metal compounds (SA@MX)SACs supported by metal compounds include carbides, nitrides, sulfide and oxide. The SAC with transition metal carbides acts momentous part in regulating the catalytic process. For their potential applications in fuel cells, there is a great need to farther improve their intrinsic activity and stability by optimizing geometric and electronic effects. Wang et al. methodically considered the effects of TMCs as substrates on CO2RR on supported Pd catalysts by combining model surface studies, real catalyst studies, in situ characterization and DFT calculations [60]. The experimental results showed that the TMCs substrate played the predominant role on the CO2RR process, thus single Pd atom exhibited the best CO2RR activity and TaC showed good stability as a substrate. Compared to commercial Pd/C, Pd/TaC exhibited the superior CO2RR activity but the lower Pd content. Lee et al. prepared a Pt/TiN catalyst on the titanium nitride support, performing the good pathway selectivity for electrochemical oxygen reduction reactions [62]. In recent years, two-dimensional (2D) MoS2 crystals with fascinating physical and chemical properties have drew increasing focus in many research fields [75-77]. In the hydrogen evolution reaction (HER), two-dimensional (2D) MoS2 is considered to be a potential substitution for Pt-based catalysts. Nonetheless, the catalytic activity of 2D MoS2 always comes from the edge position, making a great number of in-plane regions inert. Xinhe Bao's team researched that the catalytic activity of the in-plane S atom of MoS2 could be triggered by the doping of single metal atoms for HER [71]. From Fig. 2b, the HAADF-STEM image of Pt-MoS2 shows the individual dispersion of Pt atoms (marked by the red circles). Moreover, the extended X-ray absorption fine structure (EXAFS) spectra exhibited that no Pt–Pt bond exist in Pt–MoS2, further illustrating uniformly dispersed of single Pt atoms. Instead, X-ray absorption near-edge structure (XANES) and EXAFS both confirming that Pt coordinated with S. These results demonstrated that atomic Pt was immobilized in the single Mo vacancy. The right panel shows the analog geometric configuration of Pt-MoS2, and calculated adsorption free energy of H* on unsaturated S atoms in doped shells and Pt atoms in supported shells. The blue, green, yellow spheres refer to Pt, Mo, and S atoms, respectively. The results indicated that H atom would own the similar adsorption ability in both the doped and supported cases, resulting in a similar HER activity which agreed well with the HER measurements. In addition, this work elucidated that both the single Pt atom-doped layer of MoS2 nanosheets (Pt-MoS2) and Pt directly supported on 2D MoS2 (Pt/MoS2) considerably improved HER performance in contrast to pure 2D MoS2. However, Pt-MoS2 possessed a better poisoning resistance and catalytic stability than Pt/MoS2. In accordance with DFT calculation results, the regulation of HER activity could be achieved by adjusting the H atoms adsorption behavior on its neighboring S atoms by doped Pt atoms. This regulatory effect of doped Pt atoms was derived from the novel electronic states of Pt-MoS2. Besides the sulfide support, Zhang Tao et al. reported a thermally stable and easily obtained Pt SAC on the metal oxide support for the efficient methane combustion [78]. The isolated Pt atom with a high oxidation state was stabilized by the strong covalent interaction with iron and oxygen atoms on the interface. Both of the computational and experimental simulation researches have shown that the iron oxide reducibility was critical for anchoring atoms. By simply doping Pt species on the iron oxide, this no defects stabilization approach can be expanded to non-reducible supports, paving the new way for the construction of high-loading SACs for various industrial reactions.
2.1.3. Single metal atom anchored on non-metallic carbon-based support (SA@C)The SACs with the atomic metals trapped on the carbon defects of the support is depicted as the SA@C. The apparent advantages of carbon support are the low cost, easy preparation, and the excellent tolerance in the media of alkaline and acid. Zhang et al. successfully captured atomic Ni species by defective graphene (DG) named A-Ni@DG catalyst, since the defective sites are coordinated unsaturation even providing the dangling bonds for atomic metals [79]. The theoretical calculations and experiments confirmed that the excellent catalytic properties for OER and HER were originated from the specific coordination configuration of the atomic Ni. Sun et al. studied the practical synthesis of individual single Pt atoms fixed onto graphene nanosheets using atomic layer deposition (ALD) techniques [80]. The single atom catalyst exhibited significant boosted catalytic activity (up to 10 times) compared to the advanced commercial Pt/C catalysts. Atomic metals anchored on the pure carbon support achieves the low cost and easy preparation, and more importantly the practical application prospects are broad, such as energy conversion [81], fuel cells [82]. Recently, a new type of nitride porous graphene C2N has attracted attention. It can be successfully prepared through an uncomplicated bottom-up wet chemistry reaction [83]. The C2N layer consists uniformly distributed holes, terminated by sp2-bonded nitrogen atoms. And the C2N layer shows superior thermal stability in the air under 700 ℃ as well as the relatively high electrical conductivity due to the delocalized π* electron around the carbon nitride matrix. Javeed Mahmood et al. obtained the Fe@C2N catalyst by in-situ embedding the Fe3+ precursor into C2N [72]. Reduction of Fe3+ resulted in the formation of Fe oxide (FexOy) nanoparticles, which were simultaneously transformed into high crystallinity Fe° nanoparticle cores, while C2N was catalyzed into well-defined graphite nitride shells (Fe@C2N nanoparticles) during heat treatment. After leaching with 4 mol/L HCl, the Fe@C2N catalyst was obtained. The Fe@C2N exhibited the magnificent ORR activity much superior to the commercial Pt/C in both acidic and alkaline media. Moreover, its durability was also superior to the Pt/C catalyst. The structure of the Fe@C2N catalyst is shown in Fig. 2c, the d-spacing between the encapsulation layers (0.34 nm) matches the d-spacing of the graphite layer. In addition, C3N4 can also be a good support [84, 85], and some SACs based on C3N4 have been reported in details [86], such as Pd(Pt)-C3N4 [87], showing the excellent electrocatalytic performances. Li et al. studied that single Mo atom supported on N-doped carbon performed the remarkable HER performance [88]. It is worth noting that compared to Mo2C and MoN, Mo1N1C2 catalyst presented a better activity due to the unique active site structure of the Mo1N1C2 moiety exhibited by the density functional theory (DFT) calculations. It is very important for the synthesis of Mo based HER catalyst with high activity and stability.
2.1.4. Single metal atom anchored on MOF (SA@MOF)Metal organic frameworks (MOFs) is a new porous crystal [89, 90], which is a group of organic linkers and metal-containing nodes. The high porosity of the MOF allows the active site to be exposed/approached for catalysis and facilitates substrate/product transport [91, 92]. MOFs can be used as support materials. As we all known, for precious metal nanoparticles, improving the catalytic activity by supporting these metal nanoparticles or atoms on a carrier material with a large BET surface area is a cost-effective way because the rarity and high cost of noble-metal catalysts [93, 94]. At the same time, the surface electronic and geometric characteristics and morphology of metal nanoparticles can be adjusted by the support material, which is closely related to the catalytic activity [95, 96]. More importantly, MOFs can offer a variety of different chelating coordination sites through rational synthesis to stabilize individual metal atoms [97-101]. Xinzuo Fang et al. successfully loaded the single Pt atom in a MOF, exhibiting the remarkable activity for the photocatalytic water splitting due to the strong charge polarization that electrons were transferred from the MOF photosensitizer to the Pt acceptor [102]. Deserve to be mentioned, the atomic Pt performs a turnover frequency as high as 35 h-1, approximate 30 times than that of Pt nanoparticles stabilized by the same MOF. Density function theory (DFT) calculations further demonstrated that the introduction of Al-TCPP (A highly stable aluminum-based porphyrinic MOF, formulated as (AlOH)2H2TCPP (H2TCPP = 4, 4', 4", 4"'-(porphyrin-5, 10, 15, 20-tetrayl) tetrabenzoate) to the Pt SAC not only prompted the density of states near the Fermi level, but also increased the hydrogen binding energy. Yadong Li et al. used a MOF assisted strategy to synthesize a catalyst containing a single Ni site and achieved a highly efficient electroreduction of CO2 [73]. The art of synthesis is the strict mass ratio of the Zn ions and Ni ions, since the authors believe that the abundant Zn ions around the Ni are capable to prevent the Ni atoms from aggregation during the pyrolysis. The aberration-corrected HAADF-STEM image of Ni SAs/N-C shows that the Ni atoms are atomically dispersed, and the schematic illustration of Ni SAs/N-C is shown in Fig. 2d. It exhibited a distinguished turnover frequency for the electroreduction of CO2 up to 5273 h-1. Furthermore, at the overpotential of 0.89 V, the Faraday efficiency of the CO yield exceeds 71.9%, and the current density is 10.48 mA/cm2.
2.1.5. Single metal atom anchored on zeolite (SA@ZIF)The excellent microporous channels and thermal stability make zeolite an excellent support to stabilize single atom [103-106]. And the most unique is that zeolite coated metal catalysts have special and excellent shape selection catalytic performance compared to other solid supports, which also makes them very attractive in the field of catalysis [107]. As shown in Fig. 2d, Qiming Sun et al. synthesized the Rh atom catalyst within the MFI-type silicalite-1 (S-1) using in-situ hydrothermal method and hydrogen reduction in the reaction gel [74]. In view of the three-dimensional properties of zeolite crystals, they adopted different crystallographic orientations to observe the clear position of Rh. As shown in Fig. 2e, in the (011) direction, the projections are very dense, so it is impossible to distinguish atomic columns or even member rings; only the "rectangular units" marked by red circles can be recognized. Since these units consist only of sinusoidal 5-membered rings (5-MRs), the differences observed there can only be attributed to the presence of Rh species confined within these rings. Moreover, in the model shown in Fig. 2e, the marked yellow rectangular units correspond to the 5-MRs along (011) and (010). X-ray absorption spectroscopy (XAS) results prove that Rh is atomically dispersed and stabilized by oxygen atoms in the zeolite framework. The performance test results of the catalyst show that at 298 k, the hydrolysis rate of ammonia borane (AB) to generate H2 is 699 min-1. At the same time, the catalyst also exhibits admirable catalytic activity for the shape-selective hydrogenation reaction of a variety of nitroaromatics coupled AB hydrolysis, and the corresponding amine product yield reaches 99%.
2.2. PreparationThe preparation of SAC is an important factor that limiting its development. Currently, strategies for preparing SAC have been widely reported [32, 34, 88, 108-114]. They can be divided into two categories approximately: Bottom-up and top-down. The bottom-up approach is to adsorb, reduce and anchor the metal precursor on the carrier, including coprecipitation [115], adsorption [116], photochemistry [117], atomic layer deposition [118], and the like. However, excessive metal precursors lead to the agglomeration of metal atoms. In addition, although there are many types of defect sites for supporting single atoms, it is hard to accurately and controllably synthesize SAC. Therefore, it is particularly important to optimize the synthesis steps.
The bottom-up strategy [119] including (1) Atomic layer deposition (ALD) [120]. The advantage of this strategy is its precisely controllable deposition parameters. Particularly, the remarkable uniformity, the repeatability of deposition, and the controllable growth of different materials with different morphologies in the atomic layer, prove ALD to be an ideal fabricating strategy for the basic research of SAC. Atomic layer deposition instrument is shown in Fig. 3a. It consists of precursor supply system, reactor system, vacuum system, control system. These systems allow gas phase precursor pulses to alternately pass into the reactor, and chemically adsorb and react on the deposition substrate to form a deposited film. Therefore, ALD is a gas phase method based on sequential, self-limiting surface reactions. So that when the surface species may decompose and allow additional adsorption the stability of the catalyst could be low. Considering the low yield, as well as its expensive equipment, the use of this technology for the preparation of commercial SACs is currently limited. (2) Mass-selected soft-landing [121-123]. Size selection cluster deposition device as shown in Fig. 3b, and it is suitable for any type of planar support, since this is a physical deposition method. However, it has a disadvantage in exquisite experimental conditions (experiment requires an ultra-vacuum environment) and low yield. The deposition approach is not accessible for mesoporous material supports with a high specific surface area. (3) Wet-chemistry method. Fig. 3c clearly exhibits the preparation of a single metal atom embedded in a nitrogen-doped porous graphene framework (M-NHGF, M=Ni, Co, and Fe) by wet chemistry methods [109]. Briefly, aqueous suspensions of graphene oxide (GO), H2O2, and metal precursors were synthesized in corresponding proportions and hydrothermally treated to self-assemble into 3D graphene hydrogels. After lyophilization, the gel was then thermally annealed in an NH 3 atmosphere to obtain M-NHGF. Experimental results showed that Ni-NHGF was a highly active and stable O2 evolution reaction (OER) catalyst. The benefit of this approach is that no special equipment is required and it can be implemented in a conventional chemical laboratory. Moreover, SACs can be readily obtained by ultralow metal loading, despite the low loading amount is detrimental to practical catalytic applications. Co-precipitation is a kind of wet chemistry method referring to a method for synthesizing a supported metal catalyst by adding a precipitating agent to a solution containing two or more cations, and obtaining a uniform dispersion of different active species through a precipitation reaction. In the research of electrocatalysis, the onset of reaction (definitized by the onset potential) could be triggered by a low metal loading amount. However, the current density usually determined by many parameters in the catalyst preparation process which need to be strictly controlled. These factors include the adding rate of the precursor, droplet size, stirring degree, reaction temperature, pH value and reaction time etc. [124-127]. (4) Photo-assisted synthesis. The photochemical reduction assisted method is an effective means for synthesizing SACs and achieving the high loading amounts. Nanfeng Zheng's team prepared Pd1/TiO2 catalyst by photo-assisted synthesis [117]. They added H2PdCl4 as a precursor in an aqueous solution of TiO2. The entire solution system was irradiated under the ultraviolet light of a Xe lamp for ten minutes, and then the Pd1/TiO2 catalyst was obtained by centrifugal washing. Mechanism study found that ethylene glycol adsorbed on the surface of TiO2 could act as a reducing agent, generating free radicals under ultraviolet light and reducing Pd2+ to metal Pd atoms. This synthesis method makes the metal Pd reach a single atom loading amount as high as 1.5 wt%, satisfying the SAC for the future practical large-scale deployment. (5) Electrochemical deposition. The synthesize of metal SACs by electrochemical deposition method was proposed in 2018 [128]. This method is applicable to a broad territory of metals and supports for the synthesize of SAC. The deposition is executed on the cathode and anode at the same time, in which different redox reactions make SAC have different electronic states. Zhirong Zhang et al. proposed that the SAC deposited on the cathode shows high activity for the HER, while the SAC deposited on the anode shows high activity for the ORR [129]. The key of the electrochemical deposition strategy is to dominate the deposition rate of metal atoms and the number of carrier loading sites. Too fast or insufficient loading sites will induce the cluster of metal atoms.

|
Download:
|
| Fig. 3. (a) Atomic layer deposition instrument. (b) Size selection cluster deposition device. Preparation route of (c) M–NHGFs. Reproduced with permission [108]. Copyright 2018, Springer Nature. (d) Co SAs/N-C. Reproduced with permission [34]. Copyright 2016, Wiley-VCH. (e) Fe-ISAs/CN. Reproduced with permission [109]. Copyright 2017, Wiley-VCH. | |
{kind=link}
With respect to the top-down strategy, it uses nanoparticle materials or larger-scale elemental substances as precursors to achieve the conversion of single atom sites under specific conditions. It is based on breaking up ordered nanostructures into smaller pieces to provide the required performance [129]. Strong metal-support interaction acts momentous part in the stabilization and even redispersion process of top-down strategy. Rui Lang et al. found that single metal atoms can be fixed by strong covalent metal-support interaction (CMSI) independent of surface defects [78]. Even in the case of low surface area, high concentration of thermally stable single atom materials can be generated. Specifically, the high oxidation state of individualed Pt atoms are not immobilized by surface defects, but by the CMSI with iron and oxygen atoms on the surface. Experimental and DFT studies show that the reducibility of metal oxides determines the ability of the carrier to imbed the separated Pt atoms: α-Fe2O3 advocates atom-dispersed Pt, while Al2O3 advocates NP sintering. This defect free stabilization strategy can be generalized to non-reducible supports via commonly doping ferric oxide, which opens up a new way to construct highly loaded SAC for various important industrial catalytic reactions. Similarly, Kaipeng Liu et al. prepared thermally stable Ru SACs directly from commercial RuO2 powders by heating the physical mixture of RuO2 and strongly interacting supports [130]. Strong CMSI with MgAl1.2Fe0.8O4 promotes the transformation of RuO2 powder into isolated Ru atoms. Specifically, the strong CMSI between Ru and Fe plays a crucial role not only in capturing and stabilizing ruthenium atoms, but also in promoting the maturation of RuO2 aggregates. This simple and low-cost synthesis method lays a foundation for extensive production of thermally stable SAC with high metal loading. In addition, in their other work, it was also proved that SACs could be prepared by strong covalent CMSI [131]. In this study, they used iron modified spinel as support, and successfully prepared highly supported and thermally stable Pt SAC by using the strong interaction between FeOx species and Pt. The above research works show that the strong CMSI proves effective in stabilizing the single atom to prepare high activity and high stability catalysts, and also contribute a contemporary idea for the preparation of SACs. Top-down strategy including (1) MOF assisted synthesis. Peiqun Yin et al. successfully prepared Co SAs/N-C catalyst, as shown in Fig. 3d, during the annealing process (under N2 environment), the organic linker of the MOF generates N-doped porous carbon through a pyrolysis process [34]. The metal nodes are then reduced by the generated carbon at high temperatures. The reason why the Co-Co bond is not formed at high temperature is that the pre-precipitated Zn mixture changes the distance between adjacent Co atoms and contributes more free N sites. Finally, by evaporating low-boiling Zn atoms, the remaining Co atoms are anchored on the N-doped carbon support. Notably, the acquired Co-Nx single site exhibited excellent ORR performance, and the half-wave potential (0.881 V) was more positive than the commercially available Pt/C (0.811 V).
According to the durability test results, during the electrocatalytic process a single Co atom exhibited excellent chemical stability, and also remarkable thermal stability (it could withstand sintering at 900 ℃). However, a major problem regarding the experimental method is the reaction conditions which is required to be strictly controlled. These conditions usually include the reaction temperature, the choice of solvent, the ratio of the metal ion to the ligand and its concentration, and the like. Small changes in them can lead to difference in crystal quality and yield, and even new structures [73, 99, 132-138]. For example, if the reaction temperature is too low during the synthesis process, the metal ions and ligands would not cross the energy barrier and be coordinated, or the temperature is too high, the MOF structure of the product may be disrupted. In addition, the coordination capacity of ligands varies greatly at different temperatures. Differences in ligand coordination capabilities can lead to differences in the resulting MOF structure. (2) High-temperature pyrolysis strategy [139]. As we can see in Fig. 3e, Li et al. successfully separated and encapsulated an iron(Ⅲ) acetylacetonate (Fe(acac)3) by using a molecular-scale cavity of the zeolitic imidazolate skeleton (ZIF-8) as a host [109]. Iron(Ⅲ) acetylacetonate (Fe(acac)3) encapsulated in a cavity without being released mainly due to the molecular diameter of Fe(acac)3 (dm = ca. 9.7 Å) between ZIF-8 pore size (dp = 3.4 Å) and cage diameter (dc = 11.6 Å). Assembly of Zn2+ and 2-methylimidazole with a Fe(acac)3 molecule to form a molecular-level cage (Fe(acac)3@ZIF-8). After heating to 900 ℃ in an Ar atmosphere, ZIF-8 was pyrolyzed into nitrogen-doped porous carbon. Simultaneously, Fe(acac)3 was reduced by carbonization of the organic linker to form an individual single iron atom (Fe-ISA/CN) fixed on the nitrogen species. One major drawback of this approach occurs that elevated temperature treatment may cause the high energy consumption, which would increase the production cost.
3. Characterization of SACsThe characterization of SAC usually refers to various advanced and precise equipment and techniques, such as synchrotron radiation and abreaction corrected scan transmission electron microscope (AC-STEM). The rare inventory of such expensive equipment in the past impeded and delayed the identification and the development of SAC. Fortunately, during the recent years, more and more precise equipment has been applied into exploring the configuration and bonding information of the atomic metals, apparently deepening the comprehension of the nature of SAC. In this part, we will summarize the characterization methods from two aspects, including the morphology and the coordination structure, to further understand the single atom catalyst.
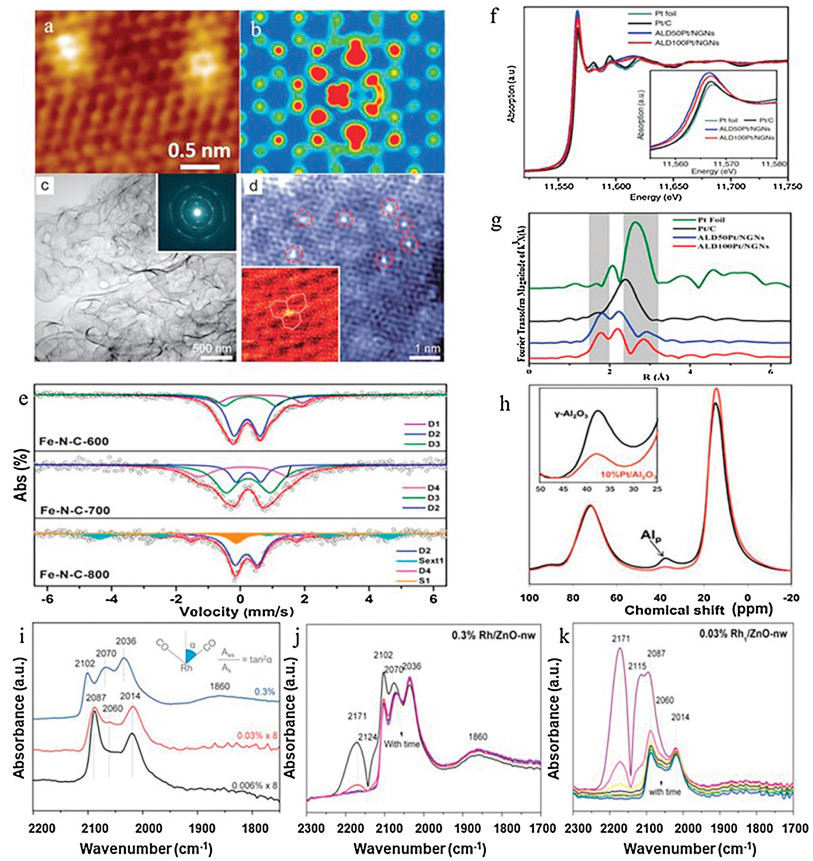
To investigate the morphology of SAC, the resolution of the microscope should at last reach 0.1 nm, otherwise the single atom and the nano cluster could not be properly distinguished. Herein, the scanning tunnel microscope (STM) and AC-STEM are widely utilized in the study of SAC. Bao and co-workers applied STM to investigate the highly exposed and doped copper(I)-nitrogen (Cu(I)-N) active sites in the graphene matrix [132].Fig. 4a shows an atomic-resolution STM image of a Cu-N site embedded in graphene, with the apparent bright spot observed at the copper center, indicating that the Cu atoms are atomically dispersed. Meanwhile, the C and N atoms around the Cu atoms are also electronically rich. The STM simulation (Fig. 4b) further reveals the atomic structure that the atomic scattered Cu-N2 center is embedded in the graphene lattice. Besides STM, AC-STEM assembled with the spherical aberration correction could characterize the local spatial resolution information of the atomic metals on the nano scale. Mingwei Chen et al. characterized the structure of Ni-doped np-G by AC-STEM [140]. The bright-field TEM image (Fig. 4c) exhibits a complex 3D morphology of np-G. Except for a few dark nickel clusters with a size of about 1-3 nm, no nickel nanoparticles were detected, confirming that the nickel template was completely dissolved. Analysis of the selected area electron diffraction (SAED; Fig. 4c, inset) shows that the irregular allocation of interconnected graphene sheets in a 3D porous structure allows np-G samples to exhibit multiple crystal orientations. Fig. 4d shows the atomic structure of Ni-doped graphene. In addition to a small amount of 1-2 nm Ni clusters, the single Ni atom displaying the significant contrast was observed. Most of the residual Ni exists as a single atom, which can be confirmed by the measured number density (about 1018 to 1019 m-2). A magnified image with atomic resolution (Fig. 4d, inset) illustrates that the single Ni species occupies the carbon sites in the graphene lattice.

|
Download:
|
| Fig. 4. (a) Atomic-resolution scanning tunneling microscopy image and (b) the corresponding simulation image of single Cu atoms embedded in nitrogen-doped graphene. Reproduced with permission [132]. Copyright 2016, Royal Society of Chemistry. (c) The TEM image of Ni-doped np-G. Inset: SAED mode. (d) HAADF-STEM image of Ni-doped graphene. Inset: An enlarged HAADF-STEM image (red circle) showing alternative Ni atoms (bright orange dots) occupying carbon sites in the graphene lattice (white lines). Reproduced with permission [133]. Copyright 2015, Wiley-VCH. (e) Fe Mössbauer spectra of the Fe-N-C-600/700/800 samples. Reproduced with permission [59]. Copyright 2017, American Chemical Society. (f) The normalized XANES spectra at the PtL3-edge of the ALDPt/NGNs, Pt/C catalysts and Pt foil. The inset shows the enlarged spectra at the PtL3-edge. (g) K3 weighted Fourier transform spectra. Spectra acquired from EXAFS of ALDPt/NGNs catalysts, Pt/C catalysts, and a Pt foil. Reproduced with permission [134]. Copyright 2016, Nature Publishing Group. (h) The27Al MAS-NMR spectra of g-Al2O3 (black) and 10 wt% Pt/g-Al2O3 (red) (both samples were calcined at 573 K before NMR measurements). Reproduced with permission [135]. Copyright 2009, American Association for the Advancement of Science. (i-k) The CO adsorption DRIFT spectra of Rh/ZnO-nw catalyst at 25 ℃ under different Rh loadings. Reproduced with permission [136]. Copyright 2016, Wiley-VCH. | |
{kind=link}
The coordination structures of the SAC are vital for its catalytic performance and the selection of reaction pathways. To disclose the configuration of the SAC, Mössbauer spectroscopy, synchrotron radiation facility and Nuclear Magnetic Resonance (NMR) Spectroscopy are mostly applied. Mössbauer spectroscopy detects small changes in the energy level of the nucleus corresponding to its environment. In general, three styles of nuclear interactions can be detected: isomer shifts, also known in the past literatures as chemical shifts; quadrupole splitting, and magnetic hyperfine splitting. At present, only 42 kinds of chemical elements (excluding post-uranium elements) with the Mössbauer effect are found. Among them, 57Fe is the most common element studied so far using this technology. Fig. 4e illustrates the Mössbauer spectra of three Fe-N-C samples [59]. Three doublet peaks fit the spectra of Fe-N-C-600 and Fe-N-C-700 well. This also illustrates the scarcity of Fe0 species in Fe-N-C-600 and Fe-N-C-700 (lack of sextet and singlet). On contrary, in the spectrum of Fe-N-C-800, not only two doublet peaks appeared, but also a singlet (S1) (corresponding to γ-Fe) and a sextet (Sext1) (corresponding to FexC species). These results indicated that Fe-N-C-600 and Fe-N-C-700 were composed only of atomic iron, while there were aggregates of Fe0 in Fe-N-C-800.
Synchrotron radiation facility offers the high dense X-ray, endowing the elucidation of the local environment of the atomic metals via the analysis of X-ray absorption spectroscopy (XAS). There are mainly two regions in the XAS spectrum, namely X-ray Absorption Near-Edge Structure (XANES) and Extended X-ray absorption fine structure (EXAFS). In terms of energy levels, XANES is generally considered to be in the range of 50 eV over the edge; EXAFS is located between 50-1000 eV on the edge. XANES is an element-specific, local bond-sensitive spectral analysis that detects the partial density of molecular vacancies. The normalized XANES spectra of the Pt L3 edges of the ALD Pt/NGN and Pt/C catalysts are shown in Fig. 4f [134]. According to the spectra we can find that the density of the vacancy state of the Pt 5d orbit can directly affect the area under the white line (WL) on the L2, 3 edges of Pt metal. For the ALD Pt/NGN and Pt/C catalysts, the intensity of Pt L3 WL showed a small difference. The variation of the Pt WL intensity amplitude at the edge of Pt L3 was regular, and it gradually increased with the order of Pt foil < Pt/C < ALD 100 Pt/NGNs < ALD 50 Pt/NGNs, indicating the Pt of ALD 100 Pt/NGNs and ALD 50 Pt/NGNs was in the oxidation state and ruled out the possibility of Pt aggregation. With aim to further unveil the fine structure of atomic metal, EXAFS is induced. The ability of EXAFS is to provide information about the atom's local environment involving the coordination numbers and the bonding lengths etc. The distance and amount of coordination between the central atom and adjacent atom can be obtained by Fourier transform (FT) analysis of the EXAFS spectrum. Wavelet transform is a developed complement to FT, which distinguishes backscattered atoms even if they overlap substantially in R space and give strong resolution in R and k spaces. For example, the Fourier transform of the drawn EXAFS region (Fig. 4g) illustrates a few main peaks. The peak obvious in the Pt foil spectrum at 2.6 Å was related to the Pt-Pt peak. And it shifted to a lower value in the Pt/C sample (2.5 Å) and was automatically attenuated in the ALD deposited sample indicating the absence of metallic Pt. As expected, the Pt-Pt peak appeared again in the spectra of ALD 100 Pt/NGNs due to the high supporting number of Pt causing the inevitable aggregation, confirming the presence of large clusters in the ALD 100 Pt/NGNs samples. The peak near 1.7 angstroms observed in the ALD deposited sample was associated with the Pt-O or Pt-C bond. Significant deviations in the Pt-O or Pt-C bond strength from the Pt-C bond and the Pt-Pt bond of the ALD sample indicated the existence of a single Pt atom in these samples. Solid-state magic-angle spinning nuclear magnetic resonance (MAS-NMR), which can be used to analyze the binding ligands and metal species in the catalytic reaction. This technique is used to research the anchoring sites of single Pt atom with a low loading. The coordination-unsaturated pentacoordinated Al3+ was verified as the anchoring sites for the single-atom Pt on the surface of the γ-Al2O3 support, and the hydrogen reverse polarization cause the enhancement of the NMR signal to be used to determine the existence of the single-atom Au. As shown in Fig. 4h, γ-Al2O3 shows three peaks at 13, 35 and 70 ppm chemical shifts [135]. The two characteristic27Al NMR peaks of γ-Al2O3 at 13 and 70 ppm represented the octahedral coordination of Al3+ (Alocta3+) and the tetrahedral coordination of Al3+ (Altetra3+), respectively. The NMR peak at 35 ppm was attributed to Al3+ pentahedral coordination (Alpenta3+), these five coordination sites were produced by high-temperature dehydration and dehydroxylation on the surface of γ-Al2O3 (here, 573 K). The 35 ppm peak intensity was greatly reduced, indicating that Pt was loaded into γ-Al2O3, and calcination on the support and at 573 K resulted in a remarkable decrease in the number of pentacoordinates of Al3+. In contrast the intensity of the 13 ppm NMR peak of Pt/ γ-Al2O3 increased, indicating that Alpenta3+ was converted to Alocta3+ (coordinated saturation: the total number of Al3+ ions remain unchanged). These results revealed that Pt atoms were bound with Alpenta3+ site on the surface of γ-Al2O3 via an oxygen bridge, thereby co-saturating these sites (five-octahedral transformation).
The excellent molecular and surface sensitivity makes infrared spectroscopy (IR) technology widely used in the characterization of catalysts [141, 142]. Compared with the above characterization methods, the most unique feature of infrared spectroscopy is that it can track the adsorption of the probe molecules on the catalyst surface, and indirectly monitor the properties and changes of the adsorption position of the probe by inferring the changes of the vibration frequency and intensity (perturbation) of the probe molecules. When using reactants as probe molecules, infrared surface sensitivity is also helpful to in-situ or operational studies under reaction conditions [143, 144]. CO and NO are the most common infrared probes for the characterization of SACs [142, 145]. CO is most commonly used in the oxide supported SACs, which is owing to its very strong infrared signal (high absorption coefficient of ν(CO) mode) and accurate sensitivity to chemical states, geometry of adsorption sites and coordination unsaturation. [146] In addition, it has superior infrared light response to most metal oxides, the instruments used are easier to available than AC-STEM and XAS in most laboratories, and it has high sensitivity to low metal loading catalyst [147]. These advantages make CO probe infrared spectroscopy (CO-IR) the most effective technology to develop and/or study oxide loaded SACs. CO-IR is mainly used to determine the existence and uniformity of isolated metal atom species in metal-metal oxide SAC. Especially in the case of small clusters/NPs and single atoms in the same catalyst, and/or when numerous single atoms exist at different sites at the same time, CO-IR will provide specific site information conveniently and economically [147]. Rui Lang et al. expressed the preparation of Rh-SAC immobilised on ZnO [136]. The Rh/ZnO-nw was characterized by in situ diffuse reflectance infrared fourier transform spectroscopy (DRIFTS). The red shift of linear Rh0-CO (2074–2070 cm-1) after helium purge (Fig. 4j) and the existence of bridged species at 1860 cm-1 in Fig. 4i provide strong evidence for the presence of Rh clusters in 0.3%Rh/ZnO nw samples. In addition, the peaks at about 2102 and 2036 cm-1 on 0.3% Rh/ZnO nw samples are related to the symmetric and asymmetric stretching of positively charged Rh(CO)2 gem-dicarbonyl species. The lack of coverage related shift at about 2060 cm-1 (Fig. 4k) for linear Rh0-CO and the lack of bridging CO indicate that the Rh atoms in 0.03% Rh1/ZnO nw are monodispersed on the ZnO support. Moreover, the angle between the two adsorbed carbonyl groups on the catalyst is different for single atom and cluster/NPs. The calculation of this angle mainly depends on the area ratio of dicarbonyl bands in gemstones, so it can be used to judge Rh single atoms and positively charged Rh clusters. As shown in the inset in Fig. 4i, the calculated angle between the carbonyl groups of 0.3% Rh/ZnO nw is 120°, and the angle between the carbonyl groups of 0.03% Rh1/ZnO nw and 0.006% Rh1/ZnO nw is nearly 96° and 94°, respectively. This work cleverly used the CO-IR method to successfully identify Rh single atoms and nanoclusters, affording an allusion for the testimony of single atoms.
4. SACs in the catalysisThe SAC presents numerous advantages as mentioned above. In the recent decade, SACs are widely applied in the electrocatalysis, especially in some energy conversion devices, such as the fuel cell and the metal-air battery. Additionally, in the chemical engineering industry, for example artificial nitrogen fixation and carbon dioxide reduction, SACs also witness a promising prospect. Here, we summarized the utilization of SAC in several half electrocatalytic reactions which are the rate determine reactions in the realistic full reaction devices.
4.1. Hydrogen evolution reactionThe HER is the half reaction of the overall water splitting reaction, which is recognized as the green energy industry of the next generation. Pt/C is currently the benchmark catalyst for HER [148-152]. However, the costliness and shortage of precious metal Pt limits the widespread use of Pt-based catalysts. In this regard, some corresponding solutions have been proposed, one of which is to reduce the consumption of Pt but retain the activity. The current single Pt atom catalyst can effectively realize this target. The other way is to exploit a low-cost and remarkable catalytic metal catalyst as the replacement of Pt-based catalyst. Non-noble metal single-atom (Co, Ni, Mo, etc.) catalysts have low coordination active sites and unique electronic structures. These characteristics also make them exhibit HER performance similar to the Pt-based catalysts [153, 154]. In the meanwhile, the metal and its support show improved interaction and charge transfer, which makes this type of catalyst exhibit significant electrochemical stability. Sun et al. presented a general preparation approach for producing individual single Pt atoms and clusters by atomic layer deposition techniques [134]. Hydrogen evolution reactions of SACs have been investigated, exhibiting the preeminently improved activity (over 37 times) and high stability compared to the advanced commercial Pt/C catalysts. According to the X-ray absorption fine structure and DFT analyses, the remarkable performance of this catalyst was mainly due to the unoccupied density of the partially 5d orbital of the Pt atom on nitrogen-doped graphene.
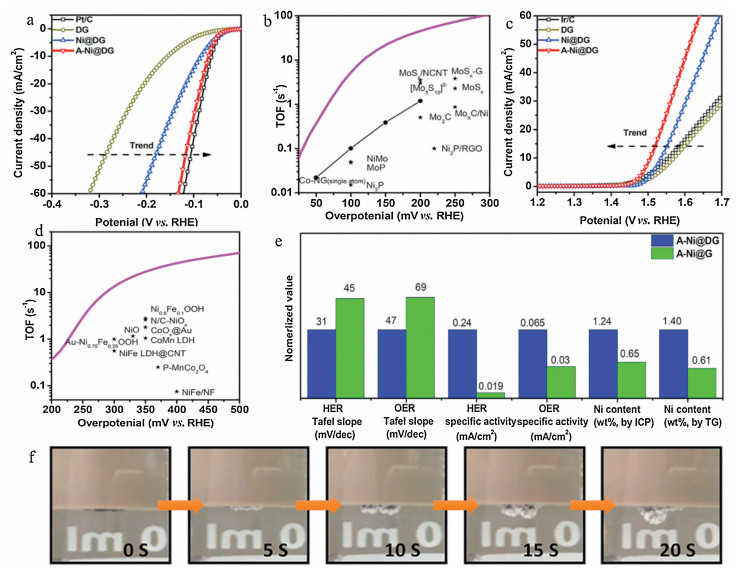
Longzhou Zhang et al. expressed a basic and inexpensive approach to produce highly stable atom-dispersed Ni catalysts (Ni loading up to 1.24 wt%) on DG(A-Ni@DG) by incipient wetness impregnation and subsequent acid leaching [79]. As illustrated in Fig. 5a, A-Ni@DG exhibits excellent HER catalytic activity, which is significantly superior to DG and Ni@DG catalysts. In Fig. 5b, the TOF value of A-Ni@DG catalyst at various overpotentials is much higher than that of the non-noble metal-based catalyst (more than ten times), confirming that the induce of Ni in the A-Ni@DG catalyst is important to promote the HER activity. And the TOF of A-Ni@DG is also superior to atomic cobalt dispersed on graphene (Fig. 5b), which confirms that atomic cobalt is worse than aNi for HER. The rapid migration of hydrogen bubbles can be observed in Fig. 5f, which also illustrates the fast kinetics of heterogeneous electron transfer in A-Ni@DG catalyst. The catalytic capability of A-Ni@DG towards OER was tested in the 1 mol/L KOH solution. As we can see in Fig. 5c that the overpotential of A-Ni@DG (270 mV) is significantly better than DG (340 mV), Ni@DG (310 mV), the reference Ir/C catalyst (320 mV) and other Ni-based catalysts at 10 mA/cm2. Impressively, A-Ni@DG has the highest TOF of these transition metal-based materials under different overpotentials of OER (Fig. 5d). The catalytic performance of A-Ni@DG for OER was significantly better than that of Ni@DG and DG. This situation confirmed that aNi is better than the bulk Ni catalysts for OER due to the tuned electronic structure of the coordination of the central Ni atom with carbon atoms. Previous studies of the coupling effects of Ni-N-C and Ni-O-C have also confirmed this rational interpretation, which enhanced oxygen release by adjusting the environment of electron at the metal-nonmetal interface, thereby promoting oxidation of OH [46].

|
Download:
|
| Fig. 5. (a) The polarization curves of DG, Ni@DG, A-Ni@DG and Pt/C were carried out in a 0.5 mol/L H2SO4 electrolyte. (b) The turnover frequency curve of A-Ni@DG and other catalysts reported in the literature for hydrogen evolution. (c) OER polarization curves of DG, Ni@DG, A-Ni@DG, and Ir/C performed in 1 mol/L KOH electrolyte. (d) The turnover frequency curve of A-Ni@DG and other catalysts reported in the literature for oxygen evolution. (e) Comparison of the Tafel slope, the specific activity obtained at a potential of 0.1 V versus RHE for HER and 1.5 V versus RHE for OER, and the Ni content obtained by ICP and TG analysis between A-Ni@DG and A-Ni@G. The heights of the A-Ni@DG bars were normalized and the heights of the A-Ni@G bars were altered accordingly. (f) A photograph of the working electrode using A-Ni@DG as a catalyst during CV measurement showed an observation of HER from 0.1 to 0.3 V for RHE. The photo is taken every 5 s. Reproduced with permission [79]. Copyright 2018, Elsevier Inc. | |
{kind=link}
For HER, the specific activity of A-Ni@DG is 0.24 mA/cm2, which is 12 times better than A-Ni@G (Fig. 5e). Similarly, for OER, A-Ni@G also performed poorly compared to A-Ni@DG. According to inductively coupled plasma and thermogravimetric analysis, there is a higher atomic Ni density in A-Ni@DG, leading to a huger number of aNi@defects on the catalyst (Fig. 5e). In this regard, the aNi@defect coordination structure has a great effect on the activity of electrocatalyst.
4.2. Oxygen reduction reactionThe oxygen reduction reaction (ORR) occurring at the cathode of the electrochemical energy device is performed by a two-electron (O2 + 2H+ + 2e- = H2O2) pathway or a four-electron (O2 + 4H + 4e- = 2H2O) pathway. In fuel cell operation, for the sake of obtaining a high efficiency, four-electron approach is preferred, whereas in the industry of organic pollution degradation the two-electron approach producing H2O2 is prioritized [155, 156]. The slow cathode ORR catalytic process and the high cost of precious metal catalysts are the main bottlenecks restricting commercial applications, just like fuel cells and zinc-air batteries. So, it is urgent to promote efficient, stable, and inexpensive ORR catalysts.
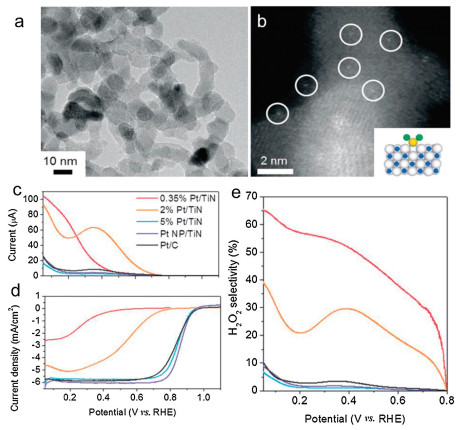
SACs not only reach the maximized atomic utilization and the adequate uncover of active sites, but also trigger the effects of strong interactions between single atoms and supports to enhance catalyst activity [157-160]. Yang et al. successfully prepared single platinum atom catalyst anchored on titanium nitride nanoparticles by means of a chlorine ligand, confirming the dual electron path for efficient and selective production of H2O2 [62]. From the TEM image of Fig. 6a, it can be seen that only TiN nanoparticles are present in the 0.35 wt% Pt/TiN sample, and Pt nanoparticles are not observed. While, the HAADF-STEM image (as shown in Fig. 6b) demonstrates the atomic Pt species (the white spots presented in the Fig. 6b). The ORR was electrochemically curried in a solution of acidic aqueous by a rotating ring disk electrode (RRDE). As we can see in Figs. 6c–e, compared with other samples, the obtained Pt SAC shows smaller disk current and larger ring current, thus confirming the high selectivity (up to 65%) of H2O2 electrocatalysis. The results illustrated that the atomic Pt species preferred the 2e- ORR pathway, whereas the Pt nano particles would like to curry the 4e- ORR pathway. In Yang's subsequent works, the effect of different support on the ORR was investigated [161]. As same as Pt/TiN SAC, an atomic Pt anchored on TiC (Pt/TiC) was prepared by just substituting the support. Compared to the counterparts of NP, no observed differences in crystalline structure, the results illustrated that Pt/TiC SAC showed more excellent catalytic selectivity, activity, and electrochemical stability for H2O2 than Pt/TiN SAC. DFT calculations indicated that the activation barrier in the 4e- path profile of Pt/TiC made the formation of H2O2 more energetic favorable. Therefore, the role of the support cannot be ignored for the development of electrocatalysis of SAC. Shaojun Guo et al. obtained a sulfur-doped Fe/N/C catalyst (expressed as Fe/SNC) by template sacrificial approach [162]. The incorporated sulfur produces a thiophene-like structure (C-S-C), reducing the electron localization near the Fe center and improving the communication with oxygen-containing species, thus promoting the 4e- path in the acidic solution. The obtained catalyst displayed a high half-wave potential (0.77 V) for ORR, and presented the Pt/C comparable stability and excellent tolerance of methanol. Recently, Guo et al. also demonstrated a metal-organic polymer supramolecule approach for the synthesis of single atom M-N-C catalysts utilizing the "self-locking" reaction between metal ions and polysaccharide, sodium alginate (SA) [163]. Fe atoms can be quaternary coordinated with N atoms on the obtained SA-Fe-N nanosheets, and they are atomically distributed. Related to the RHE, the SA-Fe-N catalyst presents remarkable ORR activity, its half-wave potential (E1/2) is 0.812 V (in 0.5 mol/L H2SO4) and 0.910 V (in 0.1 mol/L KOH), as well as excellent durability.

|
Download:
|
| Fig. 6. (a) TEM and (b) HAADF-STEM images of 0.35 wt% Pt/TiN. Rotating ring-disk electrode voltammograms of (c) ring currents concurrently obtained during the ORR with a potential maintained at 1.2 V and (d) ORR polarization curves in O2-saturated 0.1 mHClO4. (e) H2O2 selectivity. Reproduced with permission [62]. Copyright 2016, Wiley-VCH. | |
{kind=link}
The electrochemical carbon dioxide reduction reaction (CO2RR) in future renewable energy reactions has attracted much research interest as a carbon-neutral route [164-167]. The multi-proton and multi-electron CO2RR pathways competing with the hydrogen evolution reaction (HER) in aqueous solution, are sluggish in kinetics and require larger reduction bias. Therefore, it tends to exhibit low conversion efficiency and poor yield selectivity. These disadvantages impede the further evolvement and employment of CO2RR.
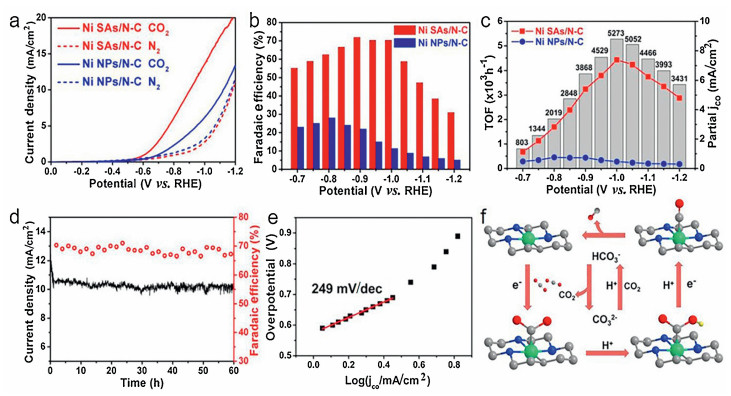
As mentioned above, Prof. Yadong Li's group utilizing metal organic frameworks (MOFs) successfully prepared SACs containing a single Ni site, endowing the efficient electroreduction of CO2 [73]. As shown in Fig. 7a, the initial potential of Ni SAs/N-C is -0.57 V vs. RHE. It is higher than the positive potential of Ni NPs/N-C. What is impressive that at -1.0 V the Ni SAs/N-C reaches a high current density amount of 10.48 mA/cm2, it is nearly three times that of Ni NPs/N-C. Working potential determines the Faraday efficiency (FE) of carbon monoxide. The maximum FE of Ni SAs/N-C reaches 71.9%, which is more than three times of Ni NPs/N-C (Fig. 7b) at a potential of -0.9 V.Fig. 7c demonstrates that, at the potential of -0.95 to -1.2 V, the partial CO current density of Ni SAs/NC is 30 times greater than that of Ni NPs/NC. Moreover, at -1.0 V Ni SAs/NC reaches the highest partial CO current density of 7.37 mA/cm2, and the TOF reaches 5273 h-1. In addition, Ni SAs/N-C density (Fig. 7d). There are two main reasons for the showed a permanent stability of 60 h without any significant attenuation of the Faradic efficiency and current significant improvement of the CO2 reduction performance of Ni SAs/N-C. One is the increase of active sites and electronic conductivity on the catalyst surface, and the other is the decrease in adsorption energy. The Tafel slope achieved by Ni SAs/N-C is 249 mV/dec (Fig. 7e), different from the bulked materials [168], indicating the diverse mechanism for reducing carbon dioxide. As on the surface of Ni SAs/N-C sufficient low-coordinated sites presents, the strong combination of CO2 may probably elucidate the distinguished performance of CO2 electroreduction to some extent (Fig. 7f).

|
Download:
|
| Fig. 7. (a) LSV curves. (b) FEs of CO and (c) partial CO current density (based on geometric surface area) plots and TOFs of Ni SAs/N-C and Ni NPs/ N-C. (d) Stability of Ni SAs/N-C. (e) Tafel plot of the partial CO current density for Ni SAs/N-C. (f) CO2 electroreduction reaction path of Ni SAs/N-C. Reproduced with permission [73]. Copyright 2017, American Chemical Society. | |
{kind=link}
Formic acid oxidation (FAOR) and methanol oxidation reaction (MOR) are two important reactions in our society [169-173]. In the past few years, the growth of suitable fuel cell systems, such as direct formic acid fuel cell (DFAFC), has gradually become the focus of attention [174-176]. However, the low kinetic rate of FAOR is a major obstacle that severely constrains the fuel cell development. Thence, the design of efficient FAOR catalysts is particularly important. The formic acid electrocatalytic oxidation mainly abides by two pathways, namely the direct pathway (the direct oxidation of formic acid to CO2) and the indirect pathway (the formic acid is first converted to the intermediate product CO and then oxidized to CO2). The indirect path produces surface poisoning substance COads with a low overpotential, thereby preventing the formic acid oxidation. In terms of the reports, the direct pathway (HCOOH → 2H+ + CO2) requires only one individual platinum atom site, while the indirect pathway (HCOOH → H2O + COads) needs a higher surface energy (based on DFT simulation) [177].
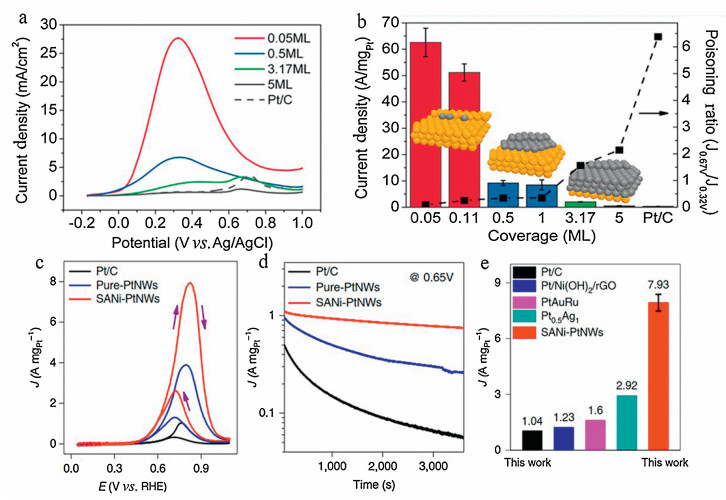
Sungeun Yang et al. systematically studied the effect of Pt coverage on the surface of the well-defined Au octahedron on the oxidation of formic acid [169]. The platinum coverage was completely regulated by a fully covered layers (5 monolayers; expressed as ML) to atomic dispersion of sub-monolayer (0.05 ML).Figs. 8a and b demonstrates the results of FAOR on samples with different coverage. The surface platinum atom specific activity indicated that the Pt atom or Pt island performed much better activity than the Pt cap layer or commercial Pt/C. The sample of 0.05 ML showed a pretty high mass activity of 62.6 A/mg. The separated Pt atoms anchored on the surface of Au (111) brought a pretty high mass activity. That was related to the lack of Pt aggregation and the bifunctional effects (both Au and Pt involved in the FAOR). The significantly enhanced mass activity of Pt islands was primarily driven by the bifunctional effects. The Pt cap layer with a great number of Pt sets and no bifunctional effect exhibited a significantly smaller mass activity for the 3.17 ML sample (2.0 A/mg) and 5 ML sample (0.52 A/mg). The poisoning rate was the current density at 0.67 V divided by the current density at 0.32 V. The large poisoning rate indicated that the FAOR occurred by an indirect route. FAOR curried on the atomic Pt species or via a direct pathway on the Pt island, whereas it abided an indirect pathway on the Pt cap layer and commercial Pt/C. The high mass activity can be guaranteed by taking control of the direct oxidation pathway without surface poisoning species, thus the Pt single atom dispersed on the Au octahedron provided the optimal activity for the direct pathway.

|
Download:
|
| Fig. 8. (a) cyclic voltammograms. (b) Comparison of the mass activities and poisoning ratios. Reproduced with permission [169]. Copyright 2013, American Chemical Society. (c) Methanol oxidation reaction (MOR) CVs. (d) Chronoamperometry MOR test. (e) The MOR peak MAs are compared to the most recent values. Reproduced with permission [173]. Copyright 2019, Springer Nature. | |
{kind=link}
Methanol oxidation reaction (MOR) proceeds through both indirect and direct routes. It is controlled by initial O-H and C-H bond activation, respectively. The direct path needs an overall size of 3–4 Pt atoms, while the secondary pathway is much less sensitive to the structure, needing only 1-2 metal atoms. Therefore, unlike formic acid oxidation, the methanol oxidation reaction is mainly controlled by an indirect path. Sun and his colleagues used ALD technology to synthesize the separated Pt atoms and assemble with grapheme [80]. The Pt SAC exhibited significantly improved catalytic activity in the MOR, its activity was 10 times higher than that of the NP counterparts. Here, Xiangfeng Duan et al. fabricated the single atomic nickel-modified Pt nanowires (SANi-Pt NWs) with abundant activated Pt sites alongside SANi and surface Pt [173]. The site is minimally blocked, thus enabling the unique design of a single Pt atom electrocatalyst with the best combination of specific activity and electrochemically active surface area (ECSA). The catalytic performance of SANi-Pt NW was evaluated using a CV with a scan rate of 20 mV/s, then under the same test conditions, the results were compared with pure Pt NW and Pt/C. The normalized CV curve of Pt mass shows a peak current density of 7.93 ± 0.45 A/mgPt near 0.816 V (vs. RHE Fig. 8c), seven times of the commercial Pt/C (1.04 A/mgPt) and twice of pure Pt NW (3.87 A/mgPt). In addition, a decline in initial overpotential of 144 mV (defined as the overpotential required to reach 0.1 A/mgPt of mass activity) was observed in SANi-Pt NW compared to Pt/C, indicating the lower activation barrier of SNAi-Pt NWs for methanol oxidation (Fig. 8c). The chronoamperometry test is applied to test the stability of SANi-Pt NW, pure Pt NW and Pt/C in MOR (Fig. 8d). It can be seen that the SANi decoration improves not only the activity but also the durability. As shown in Fig. 8e, the SANi-Pt NWs catalyst presents the highest reported specific activity and mass activity compared to the most advanced Pt or PtRu-based MOR catalysts tested under the same conductivity. It should indeed be the promising candidate catalyst for the future alkaline anion exchange membrane fuel cells.
5. Conclusion and outlooksAs the bridge to link homogeneous and heterogeneous catalysts, SACs show many fascinating features such as the maximum atom efficiency, the high stability, the selectivity and the ultrahigh specific activity. The nature of the reaction mechanism of SAC has been widely investigated mainly concentrating on the interactions between the atomic metals and the coordinated support. Although some typical reactive configurations have been proposed and elucidated, such as Me-N-C, various underlying obstacles still retain impeding the substantial comprehensions and the realistically applications of the SAC.
(1) So far, even a lot of preparation strategies of SAC have been reported, the loading amounts of the atomic metals are still not satisfied the large-scale industrial production, which is the major challenge of SAC. For the most of SACs, the loading amounts of atomic metals are normally below 2%, otherwise the metals would be inevitably aggregated. Therefrom, a fancy and convenient synthetic strategy to prepare high-loading SAC is highly desired. Jia Li et al. increased the amount of Zn atom dispersed in Zn-N-C catalyst by strictly controlling the gasification rate of Zn precursor [178]. The precursor ZnCl2 and ortho-phenylenediamine (OPD) were well converted into stable Zn-Nx active sites before gasification. Finally, the Zn-N-C catalyst with ultra-high Zn loading of 9.33 wt% was prepared. Very recently, Chao Lin et al. used the pyridine nitrogen site in a high-density conjugated organic framework material as the binding site to in situ chelate metal atoms, thereby realizing the low-temperature controllable synthesis of high-density metal single-atom catalysts [179]. The ICP-OES analysis confirmed that the metal cobalt single-atom catalyst prepared by this method can achieve a high concentration of up to 20 wt%, reaching the limit of the theoretical single-atom cobalt loading of C2N materials.
(2) The progress of SAC is closely associated with the development of the characterized techniques, such as STM, cs-TEM, XAS and Mössbauer spectroscopies. However, the scarcity of these advanced equipments restricts the broad utilization in the area of SACs. Nevertheless, more and more in-situ characterization techniques have contributed significantly to the probing of SA attributing to the advances of instrument science. Volodymyr Bon et al. reviewed recent advances in methods for the comprehensive characterization of porous skeletons using advanced in-situ diffraction and spectroscopy techniques. Just like we mentioned above, in-situ IR plays an important role in SACs characteristic. In addition, NMR spectroscopy can be used to study the mechanism of catalytic reaction in situ. Sergei S. Arzumanov et al. analyzed the conversion of ethane to aromatics on the zinc-modified zeolite BEA using 13C MAS NMR spectroscopy. [180] The reaction kinetics was monitored by1H MAS NMR at 573 K, providing information about the reaction mechanism. The kinetics of simulation experiments within the framework of the reaction kinetics scheme showed that a large amount of methane produced during the aromatization of ethane was produced by the direct hydrogenolysis of ethane.
(3) To date, all the SACs reported still could not achieve the goal of controllable synthesis. The coordination configuration of the atomic metal is not designable in the fabrication. The controllable target-oriented fabrication method should be explored to obtain the pure SAC with a specific and active coordination configuration. To this respect, the accurate ligand manipulation of SA is substantially vital to the finally coordination structures. Wanzhen Zheng et al. synthesized single-atom-level dispersed carbon nanomaterials with a metal-nitrogen coordination structure and made important progress in the electrocatalytic reduction of CO2 [181]. A series of carbon nanosheets electrocatalytic CO2 reduction catalysts with the precise structure of tetra-coordination of mono-dispersed metal atoms and pyrrole-type nitrogen atoms were synthesized by in-situ pyrolysis of metal porphyrins on the surface of melamine and cyanate polymers. Among them, the catalysts containing nickel-pyrrole nitrogen tetra-coordination structure exhibited good activity, selectivity and stability for electrocatalytic reduction of CO2 to CO.
(4) The theoretical calculations of SAC usually based on the two-dimensional models. However, the real catalysts display the three-dimensional structures, except for some single layered materials. The reaction mechanism on two-dimensional plane and three-dimensional bulk is obviously different. Therefrom, the third-dimension vertical to the surface of catalyst is recommended to take more concerns in the mechanism discussions. In addition, the current calculations are mostly established on the vacuum environment, still lacking of the situations in the liquid phase considering the fluid dynamics and mass transports. Longzhou Zhang et al. did pioneer work towards this challenge. They reported a highly efficient HER catalyst that co-trapped cobalt and platinum atoms on a hollow multilayer carbon shell, and studied the interaction between adjacent layers [182]. Theoretical studies show that the atomic metal on the inner graphite layer can significantly change the electronic structure of the outermost layer, thus adjusting the activity.
Last but not the least, it is believed that in the future, with the assistance of more innovative preparation methods, more advanced characterization tools and the in depth understanding of the catalytic mechanism especially for the role of synergetic dimers in the reaction, the goal of manipulating individual atoms and large-scale applications in the industry will be just around the corner.
AcknowledgmentsThe authors acknowledge the financial support from the National Natural Science Foundation of China (No. 22001228), "Double-First Class" University Construction Project (Nos. C176220100022 and C176220100042) and Major Science and Technology Project of Precious Metal Materials Genetic Engineering in Yunnan Province (Nos. 2019ZE001-1, 202002AB080001-6). Guangzhi Hu thanks the Double Tops Joint Fund of the Yunnan Science and Technology Bureau and Yunnan University (No. 2019FY003025).
| [1] |
Y. Ma, M. Zha, Y. Dong, et al., Mater. Res. Express 6 (2019) 115033. DOI:10.1088/2053-1591/ab45bd |
| [2] |
T. Sharifi, E. Gracia-Espino, A. Chen, et al., Adv. Energy Mater. 10 (2020) 1902084. DOI:10.1002/aenm.201902084 |
| [3] |
H. Zhang, J. He, C. Zhai, M. Zhu, Chin. Chem. Lett. 30 (2019) 2338-2342. DOI:10.1016/j.cclet.2019.07.021 |
| [4] |
S. Hu, Y. Yu, Y. Guan, et al., Chin. Chem. Lett. 31 (2020) 2839-2842. DOI:10.1016/j.cclet.2020.08.021 |
| [5] |
X. Wang, H. Gao, C. Zhai, et al., Ind. Eng. Chem. Res. 59 (2020) 19252-19259. |
| [6] |
C. Li, J. Wang, S. Feng, et al., J. Mater. Chem. A 1 (2013) 8045-8054. DOI:10.1039/c3ta11176h |
| [7] |
N. Yan, Z. Zhu, J. Zhang, et al., Mater. Res. Bull. 47 (2012) 1869-1873. DOI:10.1016/j.materresbull.2012.04.077 |
| [8] |
Y. Zhang, Y. Hu, J. Zhao, et al., J. Mater. Chem. A 7 (2019) 16364-16371. DOI:10.1039/C9TA03649K |
| [9] |
H. Yang, L. Jiang, Y. Li, et al., Nanomaterials 8 (2018) 937. DOI:10.3390/nano8110937 |
| [10] |
Q. Xie, H. Zhou, Z. Lv, et al., J. Mater. Chem. A 5 (2017) 6299-6309. DOI:10.1039/C6TA11052E |
| [11] |
Z.J. Li, A.A. Elzatahry, X. Yang, et al., Chin. Chem. Lett. 31 (2020) 1598-1602. |
| [12] |
F. Yu, Q.J. Xing, D.K. Wang, et al., Chin. Chem. Lett. 31 (2020) 1648-1653. DOI:10.1016/j.cclet.2019.08.020 |
| [13] |
J.J. Chen, Z.H. Li, Y.X. Li, Q. Yuan, Chin. Chem. Lett. 31 (2020) 1398-1401. DOI:10.1016/j.cclet.2020.03.052 |
| [14] |
J.J. Liu, Z.B. Wu, X.L. Weng, Chin. Chem. Lett. 31 (2020) 1410-1414. DOI:10.1016/j.cclet.2020.03.056 |
| [15] |
Z. Li, M. Zhu, Chem. Commun. (Camb.) 56 (2020) 14541-14552. DOI:10.1039/D0CC05709F |
| [16] |
C.N.R. Rao, M. Chhetri, Adv. Mater. 31 (2019) 1803668. DOI:10.1002/adma.201803668 |
| [17] |
J. Lai, B. Huang, Y. Chao, et al., Adv. Mater. 31 (2019) 1805541. DOI:10.1002/adma.201805541 |
| [18] |
P. Zhou, F. Lv, N. Li, et al., Nano Energy 56 (2019) 127-137. DOI:10.1016/j.nanoen.2018.11.033 |
| [19] |
P. Wang, X. Zhang, J. Zhang, et al., Nat. Commun. 8 (2017) 14580. DOI:10.1038/ncomms14580 |
| [20] |
Z.P. Xiang, H.Q. Deng, P. Peljo, et al., Angew. Chem. Int. Ed. 130 (2018) 3522-3526. DOI:10.1002/ange.201712454 |
| [21] |
Z. Wen, S. Ci, F. Zhang, et al., Adv. Mater. 24 (2012) 1399-1404. DOI:10.1002/adma.201104392 |
| [22] |
L. Xue, Y. Li, X. Liu, et al., Nat. Commun. 9 (2018) 3819. DOI:10.1038/s41467-018-06279-x |
| [23] |
S. Guo, S. Zhang, S. Sun, Angew. Chem. Int. Ed 52 (2013) 8526-8544. DOI:10.1002/anie.201207186 |
| [24] |
S. Guo, E. Wang, Nano Today 6 (2011) 240-264. DOI:10.1016/j.nantod.2011.04.007 |
| [25] |
J. Yin, Y. Li, F. Lv, et al., Adv. Mater. 29 (2017) 1704681. DOI:10.1002/adma.201704681 |
| [26] |
N. Xu, Y. Zhang, T. Zhang, et al., Nano Energy 57 (2019) 176-185. DOI:10.1016/j.nanoen.2018.12.017 |
| [27] |
M.H. Seo, M.G. Park, D.U. Lee, et al., Appl. Catal. B 239 (2018) 677-687. DOI:10.1016/j.apcatb.2018.06.006 |
| [28] |
Y. Chen, S. Hu, F. Nichols, et al., J. Mater. Chem. A 8 (2020) 11649-11655. DOI:10.1039/D0TA04633G |
| [29] |
G. Kyriakou, M.B. Boucher, A.D. Jewell, et al., Science 335 (2012) 1209-1212. DOI:10.1126/science.1215864 |
| [30] |
J. Lin, A. Wang, B. Qiao, et al., J. Am. Chem. Soc. 135 (2013) 15314-15317. DOI:10.1021/ja408574m |
| [31] |
X. Guo, G. Fang, G. Li, et al., Science 344 (2014) 616-619. DOI:10.1126/science.1253150 |
| [32] |
H. Yan, H. Cheng, H. Yi, et al., J. Am. Chem. Soc. 137 (2015) 10484-10487. DOI:10.1021/jacs.5b06485 |
| [33] |
R. Jiang, L. Li, T. Sheng, et al., J. Am. Chem. Soc. 140 (2018) 11594-11598. DOI:10.1021/jacs.8b07294 |
| [34] |
P. Yin, T. Yao, Y. Wu, et al., Angew. Chem. Int. Ed. 55 (2016) 10800-10805. DOI:10.1002/anie.201604802 |
| [35] |
X. Qiu, X. Yan, H. Pang, et al., Adv. Sci. 6 (2019) 1801103. DOI:10.1002/advs.201801103 |
| [36] |
Q. Wang, Z.L. Zhao, S. Dong, et al., Nano Energy 53 (2018) 458-467. DOI:10.1016/j.nanoen.2018.09.003 |
| [37] |
Y. Pan, S. Liu, K. Sun, et al., Angew. Chem. Int. Ed. 57 (2018) 8614-8618. DOI:10.1002/anie.201804349 |
| [38] |
Y. Qiao, P. Yuan, Y. Hu, et al., Adv. Mater. 30 (2018) 1804504. DOI:10.1002/adma.201804504 |
| [39] |
S.B. Patil, D.Y. Wang, Small 16 (2020) 2002885. DOI:10.1002/smll.202002885 |
| [40] |
J. Xu, X. Zheng, Z. Feng, et al., Nat. Sustain. 4 (2021) 233-241. DOI:10.1038/s41893-020-00635-w |
| [41] |
J. Li, Q. Guan, H. Wu, et al., J. Am. Chem. Soc. 141 (2019) 14515-14519. DOI:10.1021/jacs.9b06482 |
| [42] |
K. Jiang, S. Siahrostami, A.J. Akey, et al., Chemistry 3 (2017) 950-960. DOI:10.1016/j.chempr.2017.09.014 |
| [43] |
T. Wang, Q. Wang, Y. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 13466-13471. DOI:10.1002/anie.201907752 |
| [44] |
J. Liu, M. Jiao, B. Mei, et al., Angew. Chem. Int. Ed. 58 (2019) 1163-1167. DOI:10.1002/anie.201812423 |
| [45] |
X.F. Yang, A. Wang, B. Qiao, et al., Acc. Chem. Res. 46 (2013) 1740-1748. DOI:10.1021/ar300361m |
| [46] |
D.U. Lee, B.J. Kim, Z. Chen, J. Mater. Chem. A 1 (2013) 4754-4762. DOI:10.1039/c3ta01402a |
| [47] |
M. Yang, S. Li, Y. Wang, et al., Science 346 (2014) 1498-1501. DOI:10.1126/science.1260526 |
| [48] |
L. Zhang, J. Fischer, Y. Jia, et al., J. Am. Chem. Soc. 140 (2018) 10757-10763. DOI:10.1021/jacs.8b04647 |
| [49] |
C. Zhu, Q. Shi, S. Fu, et al., Adv. Mater. 28 (2016) 8779-8783. DOI:10.1002/adma.201602546 |
| [50] |
S. Guo, S. Zhang, S. Sun, Angew. Chem. Int. Ed. 125 (2013) 8686-8705. DOI:10.1002/ange.201207186 |
| [51] |
H. Mistry, A.S. Varela, S. Kühl, et al., Nat. Rev. Mater. 1 (2016) 16009. DOI:10.1038/natrevmats.2016.9 |
| [52] |
H. Shang, X. Zhou, J. Dong, et al., Nat. Commun. 11 (2020) 3049. DOI:10.1038/s41467-020-16848-8 |
| [53] |
Y. Yang, M. Tan, A. Garcia, et al., ACS Catal. 10 (2020) 7884-7893. DOI:10.1021/acscatal.0c00626 |
| [54] |
X. Wang, Z. Wang, F.P. García de Arquer, et al., Nat. Energy 5 (2020) 478-486. DOI:10.1038/s41560-020-0607-8 |
| [55] |
X. Wei, D. Zheng, M. Zhao, et al., Angew. Chem. Int. Ed. 59 (2020) 14639-14646. DOI:10.1002/anie.202006175 |
| [56] |
Y. Qu, B. Chen, Z. Li, et al., J. Am. Chem. Soc. 141 (2019) 4505-4509. DOI:10.1021/jacs.8b09834 |
| [57] |
X. Wang, Z. Chen, X. Zhao, et al., Angew. Chem. Int. Ed. 57 (2018) 1944-1948. DOI:10.1002/anie.201712451 |
| [58] |
X. Wang, Y. Jia, X. Mao, et al., Adv. Mater. 32 (2020) 2000966. DOI:10.1002/adma.202000966 |
| [59] |
W. Liu, L. Zhang, X. Liu, et al., J. Am. Chem. Soc. 139 (2017) 10790-10798. DOI:10.1021/jacs.7b05130 |
| [60] |
J. Wang, S. Kattel, C.J. Hawxhurst, et al., Angew. Chem. Int. Ed. 58 (2019) 6271-6275. DOI:10.1002/anie.201900781 |
| [61] |
T. Zheng, K. Jiang, N. Ta, et al., Joule 3 (2019) 265-278. DOI:10.1016/j.joule.2018.10.015 |
| [62] |
S. Yang, J. Kim, Y.J. Tak, et al., Angew. Chem. Int. Ed. 55 (2016) 2058-2062. DOI:10.1002/anie.201509241 |
| [63] |
L. Zhao, Y. Zhang, L.B. Huang, et al., Nat. Commun. 10 (2019) 1278. DOI:10.1038/s41467-019-09290-y |
| [64] |
I. Yati, M. Ridwan, G.E. Jeong, et al., Catal. Commun. 56 (2014) 11-16. DOI:10.1016/j.catcom.2014.06.016 |
| [65] |
R. Gholami, M. Alyani, K.J. Smith, Catalysts 5 (2015) 561-594. DOI:10.3390/catal5020561 |
| [66] |
W. Liu, L. Zhang, W. Yan, et al., Chem. Sci. 7 (2016) 5758-5764. DOI:10.1039/C6SC02105K |
| [67] |
N.J. O'Connor, A.S.M. Jonayat, M.J. Janik, T.P. Senftle, Nat. Catal. 1 (2018) 531-539. DOI:10.1038/s41929-018-0094-5 |
| [68] |
H.Y. Zhuo, X. Zhang, J.X. Liang, et al., Chem. Rev. 120 (2020) 12315-12341. DOI:10.1021/acs.chemrev.0c00818 |
| [69] |
J. Jones, H. Xiong, A.T. DeLaRiva, et al., Science 353 (2016) 150-154. DOI:10.1126/science.aaf8800 |
| [70] |
H. Zhang, T. Watanabe, M. Okumura, et al., Nat. Mater. 11 (2012) 49-52. DOI:10.1038/nmat3143 |
| [71] |
J. Deng, H. Li, J. Xiao, et al., Energy Environ. Sci. 8 (2015) 1594-1601. DOI:10.1039/C5EE00751H |
| [72] |
J. Mahmood, F. Li, C. Kim, et al., Nano Energy 44 (2018) 304-310. DOI:10.1016/j.nanoen.2017.11.057 |
| [73] |
C. Zhao, X. Dai, T. Yao, et al., J. Am. Chem. Soc. 139 (2017) 8078-8081. DOI:10.1021/jacs.7b02736 |
| [74] |
S. Qiming, N. Wang, T. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 18570-18576. DOI:10.1002/anie.201912367 |
| [75] |
X. Li, W. Zhong, P. Cui, et al., J. Phys. Chem. Lett. 7 (2016) 1750-1755. DOI:10.1021/acs.jpclett.6b00096 |
| [76] |
M. Xu, T. Liang, M. Shi, H. Chen, Chem. Rev. 113 (2013) 3766-3798. DOI:10.1021/cr300263a |
| [77] |
W. Zhang, Y. Zhao, K. He, et al., Nano Res. 11 (2018) 2046-2057. |
| [78] |
R. Lang, W. Xi, J.C. Liu, et al., Nat. Commun. 10 (2019) 234. DOI:10.1038/s41467-018-08136-3 |
| [79] |
L. Zhang, Y. Jia, G. Gao, et al., Chemistry 4 (2018) 285-297. DOI:10.1016/j.chempr.2017.12.005 |
| [80] |
S. Sun, G. Zhang, N. Gauquelin, et al., Sci. Rep. 3 (2013) 1775. DOI:10.1038/srep01775 |
| [81] |
K.S. Novoselov, D. Jiang, F. Schedin, et al., Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 10451. DOI:10.1073/pnas.0502848102 |
| [82] |
L. Tang, Y. Wang, Y. Li, et al., Adv. Funct. Mater. 19 (2009) 2782-2789. DOI:10.1002/adfm.200900377 |
| [83] |
J. Mahmood, E.K. Lee, M. Jung, et al., Nat. Commun. 6 (2015) 6486. DOI:10.1038/ncomms7486 |
| [84] |
M. Liang, T. Borjigin, Y. Zhang, et al., ACS Appl. Mater. Interfaces 10 (2018) 34123-34131. DOI:10.1021/acsami.8b09455 |
| [85] |
X. Xiao, Y. Gao, L. Zhang, et al., Adv. Mater. 32 (2020) 2003082. DOI:10.1002/adma.202003082 |
| [86] |
B. Yu, H. Li, J. White, et al., Adv. Funct. Mater. 30 (2020) 1905665. DOI:10.1002/adfm.201905665 |
| [87] |
G. Gao, Y. Jiao, E.R. Waclawik, A. Du, J. Am. Chem. Soc. 138 (2016) 6292-6297. DOI:10.1021/jacs.6b02692 |
| [88] |
W. Chen, J. Pei, C.T. He, et al., Angew. Chem. Int. Ed. 56 (2017) 16086-16090. DOI:10.1002/anie.201710599 |
| [89] |
D. Yang, B.C. Gates, ACS Catal. 9 (2019) 1779-1798. DOI:10.1021/acscatal.8b04515 |
| [90] |
Z. Li, H. Zhang, Q. Zha, et al., Microchim. Acta 187 (2020) 526. DOI:10.1007/s00604-020-04524-z |
| [91] |
H.C. Zhou, J.R. Long, O.M. Yaghi, Chem. Rev. 112 (2012) 673-674. DOI:10.1021/cr300014x |
| [92] |
Q.L. Zhu, Q. Xu, Chem. Soc. Rev. 43 (2014) 5468-5512. DOI:10.1039/C3CS60472A |
| [93] |
P. Falcaro, R. Ricco, A. Yazdi, et al., Coord. Chem. Rev. 307 (2016) 237-254. DOI:10.1016/j.ccr.2015.08.002 |
| [94] |
P.Z. Li, K. Aranishi, Q. Xu, Chem. Commun. (Camb.) 48 (2012) 3173-3175. DOI:10.1039/c2cc17302f |
| [95] |
B. Li, H.M. Wen, Y. Cui, et al., Adv. Mater. 28 (2016) 8819-8860. DOI:10.1002/adma.201601133 |
| [96] |
Q. Yang, Q. Xu, H.L. Jiang, Chem. Soc. Rev. 46 (2017) 4774-4808. DOI:10.1039/C6CS00724D |
| [97] |
K. Manna, T. Zhang, W. Lin, J. Am. Chem. Soc. 136 (2014) 6566-6569. DOI:10.1021/ja5018267 |
| [98] |
S. Haubenreisser, T.H. Wöste, C. Martínez, et al., Angew. Chem. Int. Ed. 128 (2016) 422-426. DOI:10.1002/ange.201507180 |
| [99] |
X. Li, R. Van Zeeland, R.V. Maligal-Ganesh, et al., ACS Catal. 6 (2016) 6324-6328. DOI:10.1021/acscatal.6b01753 |
| [100] |
H. Zhao, L. Sheng, L. Wang, et al., Energy Stor. Mater. 33 (2020) 360-381. DOI:10.1016/j.ensm.2020.08.028 |
| [101] |
X.J. Bai, X.Y. Lu, R. Ju, et al., Angew. Chem. Int. Ed. 60 (2021) 701-705. DOI:10.1002/anie.202012354 |
| [102] |
X. Fang, Q. Shang, Y. Wang, et al., Adv. Mater. 30 (2018) 1705112. DOI:10.1002/adma.201705112 |
| [103] |
L. Liu, U. Díaz, R. Arenal, et al., Nat. Mater. 16 (2017) 132-138. DOI:10.1038/nmat4757 |
| [104] |
J. Shan, M. Li, L.F. Allard, et al., Nature 551 (2017) 605-608. DOI:10.1038/nature24640 |
| [105] |
J. Lu, C. Aydin, N.D. Browning, B.C. Gates, Angew. Chem. Int. Ed. 51 (2012) 5842-5846. DOI:10.1002/anie.201107391 |
| [106] |
C. Dai, A. Zhang, M. Liu, et al., Adv. Funct. Mater. 25 (2015) 7479-7487. DOI:10.1002/adfm.201502980 |
| [107] |
D. Farrusseng, A. Tuel, New J. Chem. 40 (2016) 3933-3949. DOI:10.1039/C5NJ02608C |
| [108] |
H. Fei, J. Dong, Y. Feng, et al., Nat. Catal. 1 (2018) 63-72. DOI:10.1038/s41929-017-0008-y |
| [109] |
Y. Chen, S. Ji, Y. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 6937-6941. DOI:10.1002/anie.201702473 |
| [110] |
S. Abbet, A. Sanchez, U. Heiz, et al., J. Am. Chem. Soc. 122 (2000) 3453-3457. DOI:10.1021/ja9922476 |
| [111] |
X. Wang, W. Chen, L. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 9419-9422. DOI:10.1021/jacs.7b01686 |
| [112] |
S. Ji, Y. Chen, Q. Fu, et al., J. Am. Chem. Soc. 139 (2017) 9795-9798. DOI:10.1021/jacs.7b05018 |
| [113] |
M. Zhang, Y.G. Wang, W. Chen, et al., J. Am. Chem. Soc. 139 (2017) 10976-10979. DOI:10.1021/jacs.7b05372 |
| [114] |
Y. Pan, R. Lin, Y. Chen, et al., J. Am. Chem. Soc. 140 (2018) 4218-4221. DOI:10.1021/jacs.8b00814 |
| [115] |
B. Qiao, A. Wang, X. Yang, et al., Nat. Chem. 3 (2011) 634-641. DOI:10.1038/nchem.1095 |
| [116] |
J. Zhang, X. Wu, W.C. Cheong, et al., Nat. Commun. 9 (2018) 1002. DOI:10.1038/s41467-018-03380-z |
| [117] |
P. Liu, Y. Zhao, R. Qin, et al., Science 352 (2016) 797. DOI:10.1126/science.aaf5251 |
| [118] |
S.M. George, Chem. Rev. 110 (2010) 111-131. DOI:10.1021/cr900056b |
| [119] |
Y. Lin, H. Wan, D. Wu, et al., J. Am. Chem. Soc. 142 (2020) 7317-7321. |
| [120] |
Z. Song, Y.N. Zhu, H. Liu, et al., Small 16 (2020) 2003096. DOI:10.1002/smll.202003096 |
| [121] |
U. Heiz, A. Sanchez, S. Abbet, W.D. Schneider, J. Am. Chem. Soc. 121 (1999) 3214-3217. DOI:10.1021/ja983616l |
| [122] |
B. Lim, M. Jiang, P.H.C. Camargo, et al., Science 324 (2009) 1302. DOI:10.1126/science.1170377 |
| [123] |
Y. Lei, F. Mehmood, S. Lee, et al., Science 328 (2010) 224. DOI:10.1126/science.1185200 |
| [124] |
J. Gu, C.S. Hsu, L. Bai, et al., Science 364 (2019) 1091. DOI:10.1126/science.aaw7515 |
| [125] |
D. Liu, X. Li, S. Chen, et al., Nat. Energy 4 (2019) 512-518. DOI:10.1038/s41560-019-0402-6 |
| [126] |
N.S. Porter, H. Wu, Z. Quan, J. Fang, Acc. Chem. Res. 46 (2013) 1867-1877. DOI:10.1021/ar3002238 |
| [127] |
J. Mahmood, F. Li, S.M. Jung, et al., Nat. Nanotechnol. 12 (2017) 441-446. DOI:10.1038/nnano.2016.304 |
| [128] |
L. Zhang, J.M.T.A. Fischer, Y. Jia, et al., J. Am. Chem. Soc. 140 (2018) 10757-10763. DOI:10.1021/jacs.8b04647 |
| [129] |
Z. Zhang, C. Feng, C. Liu, et al., Nat. Commun. 11 (2020) 1215. DOI:10.1038/s41467-020-14917-6 |
| [130] |
K. Liu, X. Zhao, G. Ren, et al., Nat. Commun. 11 (2020) 1263. DOI:10.1038/s41467-020-14984-9 |
| [131] |
K. Liu, Y. Tang, Z. Yu, et al., Sci. China Mater. 63 (2020) 949-958. DOI:10.1007/s40843-020-1267-2 |
| [132] |
H. Wu, H. Li, X. Zhao, et al., Energy Environ. Sci. 9 (2016) 3736-3745. DOI:10.1039/C6EE01867J |
| [133] |
H.J. Qiu, Y. Ito, W. Cong, et al., Angew. Chem. Int. Ed. 54 (2015) 14031-14035. DOI:10.1002/anie.201507381 |
| [134] |
N. Cheng, S. Stambula, D. Wang, et al., Nat. Commun. 7 (2016) 13638. DOI:10.1038/ncomms13638 |
| [135] |
J.H. Kwak, J. Hu, D. Mei, et al., Science 325 (2009) 1670-1673. DOI:10.1126/science.1176745 |
| [136] |
R. Lang, T. Li, D. Matsumura, et al., Angew. Chem. Int. Ed. 55 (2016) 16054-16058. DOI:10.1002/anie.201607885 |
| [137] |
S. Liang, C. Hao, Y. Shi, ChemCatChem 7 (2015) 2559-2567. DOI:10.1002/cctc.201500363 |
| [138] |
T. Islamoglu, S. Goswami, Z. Li, et al., Acc. Chem. Res. 50 (2017) 805-813. DOI:10.1021/acs.accounts.6b00577 |
| [139] |
C. Wang, D. Wang, S. Liu, et al., J. Catal. 389 (2020) 150-156. DOI:10.1016/j.jcat.2020.05.034 |
| [140] |
H.J. Qiu, Y. Ito, W. Cong, et al., Angew. Chem. Int. Ed. 54 (2015) 14031-14035. DOI:10.1002/anie.201507381 |
| [141] |
F. Zaera, Chem. Soc. Rev. 43 (2014) 7624-7663. DOI:10.1039/C3CS60374A |
| [142] |
C. Lamberti, A. Zecchina, E. Groppo, S. Bordiga, Chem. Soc. Rev. 39 (2010) 4951-5001. DOI:10.1039/c0cs00117a |
| [143] |
B.L. Mojet, S.D. Ebbesen, L. Lefferts, Chem. Soc. Rev. 39 (2010) 4643-4655. DOI:10.1039/c0cs00014k |
| [144] |
A.J. Foster, R.F. Lobo, Chem. Soc. Rev. 39 (2010) 4783-4793. DOI:10.1039/c0cs00016g |
| [145] |
J. Liu, ACS Catal. 7 (2017) 34-59. DOI:10.1021/acscatal.6b01534 |
| [146] |
K.I. Hadjiivanov, G.N. Vayssilov, Adv. Catal. 47 (2002) 307-511. |
| [147] |
C. Asokan, L. DeRita, P. Christopher, Chin. J. Catal. 38 (2017) 1473-1480. DOI:10.1016/S1872-2067(17)62882-1 |
| [148] |
J. Theerthagiri, E.S.F. Cardoso, G.V. Fortunato, et al., ACS Appl. Mater. Interfaces 11 (2019) 4969-4982. DOI:10.1021/acsami.8b18153 |
| [149] |
S. Lin, Y. Liu, Z. Hu, et al., Nano Energy 42 (2017) 26-33. DOI:10.1016/j.nanoen.2017.10.038 |
| [150] |
M. Saquib, A. Halder, J. Solid State Chem. 262 (2018) 229-236. DOI:10.1016/j.jssc.2018.03.030 |
| [151] |
H. Jin, S. Sultan, M. Ha, et al., Adv. Funct. Mater. 30 (2020) 2000531. DOI:10.1002/adfm.202000531 |
| [152] |
C. Li, Z. Chen, H. Yi, et al., Angew. Chem. Int. Ed. 59 (2020) 15902-15907. DOI:10.1002/anie.202005282 |
| [153] |
P. Song, M. Luo, X. Liu, et al., Adv. Funct. Mater. 27 (2017) 1700802. DOI:10.1002/adfm.201700802 |
| [154] |
H. Su, P. Gao, M.Y. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 15194-15198. DOI:10.1002/anie.201809858 |
| [155] |
Y. Jiao, Y. Zheng, M. Jaroniec, S.Z. Qiao, Chem. Soc. Rev. 44 (2015) 2060-2086. DOI:10.1039/C4CS00470A |
| [156] |
Y. Wang, R. Shi, L. Shang, et al., Angew. Chem. Int. Ed. 59 (2020) 13057-13062. DOI:10.1002/anie.202004841 |
| [157] |
P. Chen, T. Zhou, L. Xing, et al., Angew. Chem. Int. Ed. 56 (2017) 610-614. DOI:10.1002/anie.201610119 |
| [158] |
Y.J. Sa, D.J. Seo, J. Woo, et al., J. Am. Chem. Soc. 138 (2016) 15046-15056. DOI:10.1021/jacs.6b09470 |
| [159] |
C. Zhu, S. Fu, J. Song, et al., Small 13 (2017) 1603407. DOI:10.1002/smll.201603407 |
| [160] |
X.X. Wang, D.A. Cullen, Y.T. Pan, et al., Adv. Mater. 30 (2018) 1706758. DOI:10.1002/adma.201706758 |
| [161] |
S. Yang, Y.J. Tak, J. Kim, et al., ACS Catal. 7 (2017) 1301-1307. DOI:10.1021/acscatal.6b02899 |
| [162] |
H. Shen, E. Gracia-Espino, J. Ma, et al., Angew. Chem. Int. Ed. 56 (2017) 13800-13804. DOI:10.1002/anie.201706602 |
| [163] |
Z. Miao, X. Wang, M.C. Tsai, et al., Adv. Energy Mater. 8 (2018) 1801226. DOI:10.1002/aenm.201801226 |
| [164] |
S. Gao, Y. Lin, X. Jiao, et al., Nature 529 (2016) 68-71. DOI:10.1038/nature16455 |
| [165] |
B.R. Sutherland, Joule 1 (2017) 643-645. DOI:10.1016/j.joule.2017.12.004 |
| [166] |
F. Li, H. Yang, W. Li, L. Sun, Joule 2 (2018) 36-60. DOI:10.1016/j.joule.2017.10.012 |
| [167] |
L. Han, S. Song, M. Liu, et al., J. Am. Chem. Soc. 142 (2020) 12563-12567. DOI:10.1021/jacs.9b12111 |
| [168] |
Q. Lu, J. Rosen, Y. Zhou, et al., Nat. Commun. 5 (2014) 3242. DOI:10.1038/ncomms4242 |
| [169] |
S. Yang, H. Lee, ACS Catal. 3 (2013) 437-443. |
| [170] |
W. Huang, H. Wang, J. Zhou, et al., Nat. Commun. 6 (2015) 10035. DOI:10.1038/ncomms10035 |
| [171] |
F. Ren, C. Wang, C. Zhai, et al., J. Mater. Chem. A 1 (2013) 7255-7261. DOI:10.1039/c3ta11291h |
| [172] |
Y.Y. Feng, L.X. Bi, Z.H. Liu, et al., J. Catal. 290 (2012) 18-25. DOI:10.1016/j.jcat.2012.02.013 |
| [173] |
M. Li, K. Duanmu, C. Wan, et al., Nat. Catal. 2 (2019) 495-503. DOI:10.1038/s41929-019-0279-6 |
| [174] |
M. Choi, C.Y. Ahn, H. Lee, et al., Appl. Catal. B 253 (2019) 187-195. DOI:10.1016/j.apcatb.2019.04.059 |
| [175] |
M. Mazurkiewicz-Pawlicka, A. Malolepszy, A. Mikolajczuk-Zychora, et al., Appl. Surf. Sci. 476 (2019) 806-814. DOI:10.1016/j.apsusc.2019.01.114 |
| [176] |
R. Wang, J. Liu, P. Liu, et al., Nano Res. 7 (2014) 1569-1580. DOI:10.1007/s12274-014-0517-9 |
| [177] |
M. Neurock, M. Janik, A. Wieckowski, Faraday Discuss. 140 (2009) 363-378. DOI:10.1039/B804591G |
| [178] |
J. Li, S. Chen, N. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 7035-7039. DOI:10.1002/anie.201902109 |
| [179] |
C. Lin, H. Zhang, X. Song, et al., Mater. Horizons 7 (2020) 2726-2733. DOI:10.1039/D0MH01061H://www.cnki.com.cn/Article/CJFDTOTAL-SYWT201903002.htm |
| [180] |
S.S. Arzumanov, A.A. Gabrienko, D. Freude, A.G. Stepanov, Solid State Nucl. Magn. Reson. 35 (2009) 113-119. DOI:10.1016/j.ssnmr.2008.12.005 |
| [181] |
W. Zheng, F. Chen, Q. Zeng, et al., Nano-Micro Lett. 12 (2020) 108. DOI:10.1007/s40820-020-00443-z |
| [182] |
L. Zhang, Y. Jia, H. Liu, et al., Angew Chem. Int. Ed. 58 (2019) 9404-9408. DOI:10.1002/anie.201902107 |