 2021, Vol. 32
2021, Vol. 32 


In the past decade, gold nanoparticles (Au NPs) have attracted great attention in various photocatalytic reactions due to their strong light converting ability in visible range and high catalytic activity even at low temperature. Examples are the reduction of aromatic nitro compounds [1-4], CO oxidation [5] and carbon-halogen bond cleavage [6]. Under resonant light irradiation, the surface plasmons on Au NP surface undergo non-radiative decay and generate hot electron-hole pairs that can promote photocatalysis. On the other hand, Au NPs are excellent thermocatalysts in conventional heterogeneous catalysis, where the Au surface activates the reactant molecules and/or changes the reaction pathway without the presence of hot carrier [7-9]. Recent studies on Au plasmonic photocatalysis mainly focus on the use of hot electrons (holes) in chemistry; however, whether the Au NPs act also as second role to change the reaction pathway of the photocatalysis remains unclear. In situ monitoring of the interfacial chemistry on Au NPs is required to understand the plasmonic reaction mechanism at molecular level.
Surface-enhanced Raman spectroscopy (SERS) is widely used to monitor catalytic reactions on the surface of noble metal NPs. The high sensitivity and molecular specificity of nanostructures enable SERS a powerful analytical method to explore interfacial reaction mechanism [10-12].
Au NPs are excellent SERS substrates because they are stabler and easier to prepare than any other plasmonic metals (Ag and Cu) [5, 13-15]. Typically, bifunctional nanostructures have to be prepared by using both plasmonic (Au or Ag) and catalytic metals (CuO, Pt or Pd) for the detection of interfacial catalysis [16-20]. Since SERS and hot carriers are both originated from plasmon decay, if the chemical reaction occurs on the plasmonic Au surface, SERS can be employed to monitor the plasmonic catalysis without using complex bifunctional nanostructures.
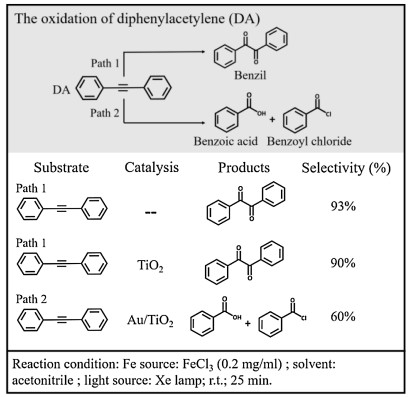
Photocatalytic alkyne oxidation reactions have significant applications in synthetic chemistry and products like epoxide, ketone and acid are important chemical materials for producing steroidal medicines. Although selective conversion of the substances to preferred products can be achieved by controlling the level of oxidation, the reaction pathway is difficult to change under a certain reaction condition. Here, we present a proof-of-concept study by using Au nanocube (NC) film as a plasmonic substrate to study diphenylacetylene (DA) oxidation as our model reaction. It was found that the plasmonic Au changes the reaction pathway via activation of C≡C and produces benzoic acid and benzoyl, instead of benzil product when Au NPs are absent. Since plasmon-induced catalysis on Au NPs is generally limited by the charge-carrier recombination, a number of Au-semiconductor hybrid nanomaterials have been employed to improve the electron-hole separation for high reactivity in photocatalysis; examples are deuteration [21], C-C coupling [22], N2 fixation [23], ethanol oxidation [24], and CO2 reduction [25-27]. Therefore, we prepared Au/TiO2 to improve the plasmonic catalytic activity in synthetic chemistry. What we found in SERS explains the increased selectivity of DA oxidation to the two products in Au/TiO2 photocatalytic system (Fig. 1). According to our experimental results, it was proposed that the hot holes play a key role in C≡C activation and both the plasmonic hot electrons and holes are involved in the photooxidation.

|
Download:
|
| Fig. 1. Reaction pathways of DA oxidation in the presence of FeCl3 in acetonitrile. | |
{kind=link}
Au NCs with a diameter of ~50 nm and Au NPs with a diameter of ~10 nm were synthesized by using sodium borohydride and ascorbic acid reduction method in a mixture of an aqueous solution of cetyltrimethyl ammonium chloride (CTAC). In the photocatalytic oxidation of DA, a Xe lamp was used (PLS-SXE300, Beijing Trusttech Co., Ltd.) as the irradiation light source. The concentrations of DA and FeCl3 are 1 and 0.2 mg/mL, respectively. More experimental details are available in the Supporting information.
In a control experiment of DA oxidation without adding Au NPs, benzil was formed after the reaction mixture was illuminated under visible light in the presence of FeCl3 (Fig. 1, Path 1). When Au NPs (supported on TiO2, see Fig. S1 in Supporting information) were added as catalyst, the concentration of benzoic acid (BA) and benzoyl chloride (BOC) in the product dramatically increased (Fig. 1, Path 2). In contrast, this change was not observed by using TiO2 as the catalyst. It is reasonable to propose that there is a different reaction pathway for light-driven DA oxidation in the presence of Au NPs.
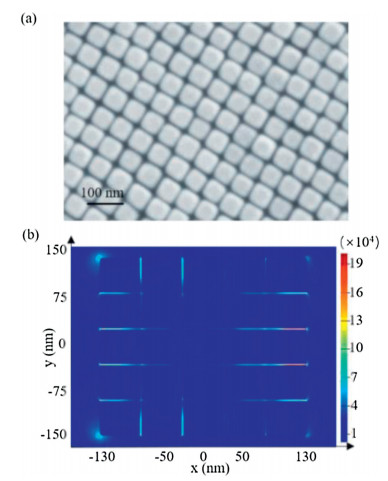
In order to monitor the molecular conversion of DA oxidation at the catalytic interfaces, we synthesized ~50 nm Au NCs (Fig. S3 in Supporting information) and then prepared closely packed nanocube film (Au NC film) as our SERS substrate. The SEM image of the as-prepared film in Fig. 2a shows the high Au NC dispersity and regular pattern formed by the NCs. The average distance of two adjacent Au NCs is about 1.85 nm, which is beneficial for strong plasmonic coupling and generation of reproducible SERS signal. According to the 3D-finite difference time domain (FDTD) simulation, the calculated enhancement factor in the gaps between the NCs is about 2×105 (Fig. 2b, and Supporting information section 1.8). For the SERS measurement, ethanethioate was linked to DA to form S-(4-(phenylethynyl)phenyl) ethanethioate (S-4-PPET) (Scheme S1 in Supporting information) with strong Au-S bonds to anchor the reactant molecules on the Au surface (Fig. 3a). In situ SERS monitoring was conducted with 633 nm laser after the Au NC film was immersed into a CH3CN solution of FeCl3. The peaks at 998 and 1140 cm−1 (Fig. 3b) are assigned to the asymmetric stretching mode of phenyl rings [19]; while the peaks at 1579 and 2216 cm−1 are attributed to the symmetric stretching mode of phenyl rings and vibration of carbon-carbon triple bonds, respectively [19, 28], which are employed to evaluate the course of oxidation reaction.

|
Download:
|
| Fig. 2. (a) SEM of Au nanocube film (Au NC film). (b) 3D-FDTD simulation of electric fields in the film. | |
{kind=link}

|
Download:
|
| Fig. 3. (a) Preparation of Au NC film coated with S-(4-(phenylethynyl)phenyl) ethanethioate (S-4-PPET). (b) SERS spectra of DA oxidation on Au NC film with FeCl3 and solvent MeCN. Blank lines are from Au NC film coated with commercial DA, BC, BA, and BOC under the same condition. (c) SERS spectra of BC on Au NC film in MeCN solution of FeCl3. * stands for peaks of MeCN. | |
{kind=link}
During the reaction, the peaks at 998, 1140 and 2216 cm−1 decrease and a new peak at ~1565 cm−1 arises gradually. According to the standard SERS spectrum of CH3S-Ph-CH2-Cl (BC, after incubation with Au substrate, Fig. 3c), the peak at 1565 cm−1 can be assigned to the symmetric stretching mode of phenyl ring of Ph-CH2-Cl analogue. And when Fe(NO3)3 is used instead of FeCl3, this peak shows a redshift of 11 cm−1 and appears at 1554 cm−1 (Fig. S4 in Supporting information), indicating the formation of a different intermediate product by changing the anions. As the reaction goes on, the peak at 1565 cm−1 disappears while the one at 1584 cm−1 arises, which is similar to that of the CH3S-Ph-CH2-Cl oxidation (Fig. 3c). The peak at ~1584 cm−1 can be assigned to the symmetric stretching mode of phenyl ring of BA and BOC (Fig. 3b).
Interestingly, the characteristic SERS signal of benzil, which was supposed to appear at around 1700−1800 cm−1 (C=O), was not observed. This is in agreement with our assumption that plasmonic Au changes the original reaction pathway: the Au surface directly induces breaking of the C≡C triple bonds without producing benzil. In addition, we noticed that Fe(Ⅲ) was reduced to Fe(Ⅱ) during the reaction process. A chemical indicator 1, 10-phenanthroline, which will give an orange coordination compound in the presence of Fe2+, shows an evidently strong absorption at ~510 nm (Fig. S5 in Supporting information) after the reaction. The change of valence might be conducted according to the following Eq. 1 under light illumination. So, it is necessary to find out whether Fe(Ⅲ)Cl3 or Fe(Ⅱ)Cl2 plays an important role in this reaction.
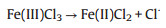
|
(1) |
As shown in Fig. S6 (Supporting information), no reaction could be detected when FeCl2 was used instead of FeCl3. As Fe3+ attaches to C≡C triple bond by dative force [29-32], the electron density of π orbital decreases and C≡C is activated for oxidation. It is also suggested that the FeCl3 to FeCl2 conversion releases Cl· [33-35] and leads to the formation of Ph-CH2-Cl analogue.
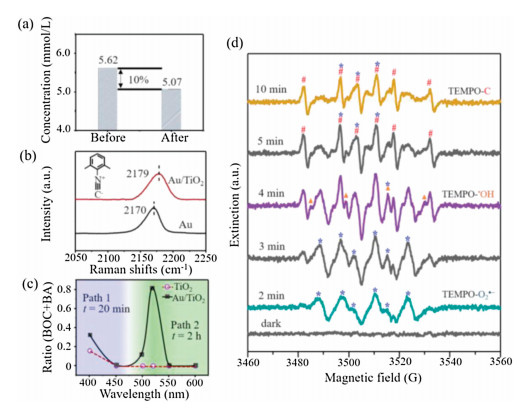
Since most photocatalytic reactions refer to adsorption and desorption processes, we further investigated whether the DA molecules have such interaction with Au NPs. First, Au/TiO2 was suspended in a CH3CN solution of DA for 40 min. After filtration, the concentration of DA was detected by GC–MS with 1-naphthol as the internal standard substance. The data displayed in Fig. 4a indicates a 10% decrease of the DA concentration. In contrast, such decrease was not observed after mixing with TiO2. It is obvious that Au NPs can adsorb DA in the reaction mixture. Many research groups have verified a similar photoadsorption of metallic nanostructures to molecules carrying C≡C [36-38]. After reaction the product molecules will leave the metal surface and the free DA in solution will approach again to keep the reaction going. Notably, a certain part of the free DA in solution will be oxidized to benzil (Path 1), causing a decrease of the selectivity of BA and BOC (Fig. 1).

|
Download:
|
| Fig. 4. (a) The concentration of DA detected by GC–MS before and after Au/TiO2 incubation. (b) SERS spectra of 2, 6-DMPI adsorbed on Au/TiO2 and Au. (c) The ratio of BA and BOC under different excitation wavelengths. (d) EPR tests of Au/TiO2 with TEMPO, DA, and FeCl3 in CH3CN under visible light irradiation; #, ▲ and * represent the peaks of TEMPO-C, TEMPO-•OH and TEMPO-O2•−, respectively. | |
{kind=link}
The interfacial charge transfer between Au and TiO2 was investigated by SERS signal of 2, 6-dimethylphenyl isocyanide (2, 6-DMPI). The peak at ~2170 cm−1 assigned to N≡C stretching will blue-shift if there is an electron donation from δ-bond of N≡C to d-band of Au, and vice versa [39]. We synthesized ~50 nm Au NPs (Fig. S1) and loaded them on TiO2 support via electrostatic forces. Before SERS detection, the Au NPs and Au/TiO2 were incubated in an alcoholic solution of 2, 6-DMPI overnight. As shown in Fig. 4b, the band shifts from 2170 (Au) to 2179 cm−1 (Au/TiO2). The blue-shift indicates an exact electron donation from Au to TiO2, resulting in a decreased electron density and efficient charge separation [40] on Au that will benefit the oxidation reaction.
The new pathway on Au NPs inspired us to explore the reaction mechanism. It was confirmed that the reaction cannot proceed without light illumination, even at elevated temperature (Fig. S2). Plasmonic hot electron-hole pairs are very active and could drive the catalytic reaction on metal surfaces [41-43]. As shown in Fig. 4c, when we compared the performance of Au/TiO2 and TiO2 at different wavelengths, the highest ratio of products BA and BOC appears at 520 nm, which is consistent with the plasmon resonance position of the Au NPs. In a negative control, Pt/TiO2 without sufficient plasmonic activity was employed and showed no catalytic performance (Fig. S7 in Supporting information). It has to be mentioned here that at 400 and 450 nm, where the DA reactant with FeCl3 shows strong light absorption, the photooxidation occurs in solution following Path 1 (Fig. 1) and generates benzil product.
In order to determine the reaction intermediates, we tested the generated radical species via electron paramagnetic resonance (EPR). A radical-trapping reagent 2, 2, 6, 6-tetramethyl-1-piperdinyloxy (TEMPO) (Fig. S8a in Supporting information) was added into the reaction mixture containing Au/TiO2, DA, FeCl3, and CH3CN. No signal was detected in the dark (Fig. 4d, black line). Under light illumination, typical EPR peaks of TEMPO-O2•− (marked with *) adducts were observed, which are generated from O2 in air reduced by the hot electrons on Au NPs [21, 44]. At the same time, TEMPO-•OH (▲) generated by trace amount of water from air and FeCl3·6H2O was detected. It has also been reported previously that a small amount of water from crystalline hydrates would be ionized in organic solvent [45]. It is noteworthy that large a amount of water will inhibit the reaction because the indispensable Fe3+ will combine with hydroxide species to form Fe(OH)3 precipitate. Furthermore, the appearance of much stronger TEMPO-C signal (marked with #) with Au/TiO2 than without (Fig. S8b in Supporting information) confirms the much easier bond breakage of C≡C on plasmonic Au NPs.
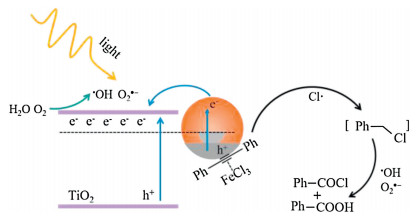
According to the results above, a reasonable mechanism is proposed in Fig. 5. Under light extinction, the plasmonic hot electrons on Au NPs are transferred to TiO2, leaving hot holes on the metal surface. The DA molecules adsorbed on Au NPs are activated by the hot holes and subsequently react with Cl· species to form Ph-CH2-Cl analogue. On the other hand, the hot electrons convert O2 and H2O to O2•− and •OH, respectively, which are active oxidants and eventually react with the Ph-CH2-Cl analogue to form the BA and BOC final products.

|
Download:
|
| Fig. 5. Proposed mechanism of DA oxidation on Au/TiO2 under visible light illumination. | |
{kind=link}
To sum up, we demonstrate that Au NPs can change the reaction pathway in DA oxidation under visible light irradiation, which results in different products of BA and BOC, instead of forming benzil without Au NPs. The highly ordered Au NC assembly provides strong electromagnetic fields for in situ SERS monitoring of the reaction. All evidences from SERS, GC-MS and EPR suggest a radical reaction pathway in which the C≡C triple bond is activated and breaks directly on the effect of hot holes on Au NPs. On the other hand, the hot electrons enable the conversion of O2 and H2O to the corresponding reactive intermediates (O2•− and •OH) and finally generate BA and BOC products. This work shows the great potential of SERS in analysing chemical reactions on metal NPs. The finding of this work discloses the second role of plasmonic NPs, in addition to the hot carrier producer, as conventional heterogeneous catalysts to interact with the molecules and change the reaction pathway.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 22022406, 21861132016 and 21775074), the Natural Science Foundation of Tianjin (Nos. 20JCJQJC00110 and 20JCYBJC00590), the Fundamental Research Funds for the Central Universities-Nankai University (No. 000082), the 111 project (No. B12015), and the National Key R&D Program (Nos. 2017YFA0206702 and 2016YFB0901502).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.014.
| [1] |
W. Xie, B. Walkenfort, S. Schlücker, J. Am. Chem. Soc. 135 (2013) 1657-1660. DOI:10.1021/ja309074a |
| [2] |
F.F. Zhang, P. Yang, K. Matras-Postolek, J. Nanosci. Nanotechnol. 16 (2016) 5966-5974. DOI:10.1166/jnn.2016.10858 |
| [3] |
J. Wei, S.N. Qin, J.L. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 10343-10347. DOI:10.1002/anie.202000426 |
| [4] |
Q.F. Zhang, Y.D. Zhou, E. Villarreal, et al., Nano Lett. 15 (2015) 4161-4169. DOI:10.1021/acs.nanolett.5b01286 |
| [5] |
H. Zhang, C. Wang, H.L. Sun, et al., Nat. Commun. 8 (2017) 15447. DOI:10.1038/ncomms15447 |
| [6] |
P. Jiang, Y.Y. Dong, L. Yang, Y.R. Zhao, W. Xie, J. Phys. Chem. C 123 (2019) 16741-16746. DOI:10.1021/acs.jpcc.9b03238 |
| [7] |
X. Liu, L. He, Y.M. Liu, Y. Cao, Acc. Chem. Res. 47 (2014) 793-804. DOI:10.1021/ar400165j |
| [8] |
D. Ren, L. He, L. Yu, et al., J. Am. Chem. Soc. 134 (2012) 17592-17598. DOI:10.1021/ja3066978 |
| [9] |
L. Luza, C.P. Rambor, A. Gual, et al., ACS Catal. 7 (2017) 2791-2799. DOI:10.1021/acscatal.7b00391 |
| [10] |
S.W. Li, P. Miao, Y.Y. Zhang, et al., Adv. Mater. 32 (2020) 2000086. DOI:10.1002/aisy.202000086 |
| [11] |
J. Liu, C.C. Zhang, S.X. Zhang, H.J. Yu, W. Xie, Chin. Chem. Lett. 31 (2020) 539-542. DOI:10.1016/j.cclet.2019.07.037 |
| [12] |
C.S.L. Koh, H.K. Lee, G.C. Phan-Quang, et al., Angew. Chem. Int. Ed. 56 (2017) 8813-8817. DOI:10.1002/anie.201704433 |
| [13] |
J.E. Park, Y. Lee, J.M. Nam, Nano Lett. 18 (2018) 6475-6482. DOI:10.1021/acs.nanolett.8b02973 |
| [14] |
Y.H. Wang, M.M. Liang, Y.J. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 11257-11261. DOI:10.1002/anie.201805464 |
| [15] |
W. Xie, S. Schlücker, Nat. Commun. 6 (2015) 7570. DOI:10.1038/ncomms8570 |
| [16] |
K.F. Zhang, L. Yang, Y.F. Hu, et al., Angew. Chem. Int. Ed. 59 (2020) 18003-18009. DOI:10.1002/anie.202007462 |
| [17] |
J.F. Huang, Y.H. Zhu, M. Lin, et al., J. Am. Chem. Soc. 135 (2013) 8552-8561. DOI:10.1021/ja4004602 |
| [18] |
W. Xie, C. Herrmann, K. Kömpe, M. Haase, S. Schlücker, J. Am. Chem. Soc. 133 (2011) 19302-19305. DOI:10.1021/ja208298q |
| [19] |
Y.R. Zhao, L.L. Du, H.X. Li, W. Xie, J. Chen, J. Phys. Chem. Lett. 10 (2019) 1286-1291. DOI:10.1021/acs.jpclett.9b00351 |
| [20] |
Y.J. Lai, L.J. Dong, R. Liu, et al., Chin. Chem. Lett. 31 (2020) 2437-2441. DOI:10.1016/j.cclet.2020.04.050 |
| [21] |
Y.Y. Dong, Y.L. Su, L.L. Du, et al., ACS Nano 13 (2019) 10754-10760. DOI:10.1021/acsnano.9b05523 |
| [22] |
E. Peiris, S. Sarina, E.R. Waclawik, et al., Angew. Chem. Int. Ed. 58 (2019) 12032-12036. DOI:10.1002/anie.201904452 |
| [23] |
J.H. Yang, Y.Z. Guo, R.B. Jiang, et al., J. Am. Chem. Soc. 140 (2018) 8497-8508. DOI:10.1021/jacs.8b03537 |
| [24] |
T.H. Tan, J. Scott, Y.H. Ng, et al., ACS Catal. 6 (2016) 8021-8029. DOI:10.1021/acscatal.6b01833 |
| [25] |
X.F. Cui, J. Wang, B. Liu, et al., J. Am. Chem. Soc. 140 (2018) 16514-16520. DOI:10.1021/jacs.8b06723 |
| [26] |
S.J. Yu, A.J. Wilson, J. Heo, P.K. Jain, Nano Lett. 18 (2018) 2189-2194. DOI:10.1021/acs.nanolett.7b05410 |
| [27] |
J.H. Yang, Y.Z. Guo, W.Z. Lu, R.B. Jiang, J.F. Wang, Adv. Mater. 30 (2018) 1802227. DOI:10.1002/adma.201802227 |
| [28] |
Y. Zeng, J.Q. Ren, A.G. Shen, J.M. Hu, ACS Appl. Mater. Interfaces 8 (2016) 27772-27778. DOI:10.1021/acsami.6b09722 |
| [29] |
W.H. Ji, Y.M. Pan, S.Y. Zhao, Z.P. Zhan, Synlett 19 (2008) 3046-3052. |
| [30] |
G. Cantagrel, B.D. Carné-Carnavalet, C. Meyer, J. Cossy, Org. Lett. 11 (2009) 4262-4265. DOI:10.1021/ol9014877 |
| [31] |
R.S. Li, S.R. Wang, W.J. Lu, Org. Lett. 9 (2007) 2219-2222. DOI:10.1021/ol070737u |
| [32] |
X.L. Bu, L.C. Hong, R.T. Liu, et al., Tetrahedron 68 (2012) 7960-7965. DOI:10.1016/j.tet.2012.07.007 |
| [33] |
H. Sugimoto, D.T. Sawyer, J. Org. Chem. 50 (1985) 1784-1786. DOI:10.1021/jo00210a053 |
| [34] |
J. Wittmer, S. Bleicher, C. Zetzsch, J. Phys. Chem. A 119 (2015) 4373-4385. DOI:10.1021/jp508006s |
| [35] |
Q. Zhao, H.M. Zhao, X. Quan, S. Chen, Y.B. Zhang, Mar. Pollut. Bull. 86 (2014) 76-83. DOI:10.1016/j.marpolbul.2014.07.040 |
| [36] |
P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi, T. Tsukuda, J. Am. Chem. Soc. 135 (2013) 9450-9457. DOI:10.1021/ja401798z |
| [37] |
G. Li, R.C. Jin, J. Am. Chem. Soc. 136 (2014) 11347-11354. DOI:10.1021/ja503724j |
| [38] |
S. Zhang, K.L. Chandra, C.B. Gorman, J. Am. Chem. Soc. 129 (2007) 4876-4877. DOI:10.1021/ja0704380 |
| [39] |
J.H. Zhong, X. Jin, L.Y. Meng, et al., Nat. Nanotechnol. 12 (2017) 132-136. DOI:10.1038/nnano.2016.241 |
| [40] |
W.J. Xu, J. Jia, T. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 22246-22251. DOI:10.1002/anie.202010613 |
| [41] |
S. Linic, U. Aslam, C. Boerigter, M. Morabito, Nat. Mater. 14 (2015) 567-576. DOI:10.1038/nmat4281 |
| [42] |
Y.C. Zhang, S. He, W.X. Guo, et al., Chem. Rev. 118 (2018) 2927-2954. DOI:10.1021/acs.chemrev.7b00430 |
| [43] |
K. Qian, E.W. Zhao, S. He, W.D. Wei, Chin. Chem. Lett. 29 (2018) 783-786. DOI:10.1016/j.cclet.2018.03.027 |
| [44] |
Z.Y. Zhang, U. Gernert, R.F. Gerhardt, et al., ACS Catal. 8 (2018) 2443-2449. DOI:10.1021/acscatal.8b00101 |
| [45] |
C. Zhou, G.L. Tian, H.Y. Shen, Y.H. Ye, Acta Chim. Sin. 59 (2001) 1707-1711. |