 2021, Vol. 32
2021, Vol. 32 


b University of Science and Technology of China, Hefei 230026, China
Ammonia (NH3) as very important chemical has been widely applied in fuel of vehicles, agricultural, plastic and textile industries [1-4]. Although abundant nitrogen (N2) exists in the atmosphere (~78%), efficient utilization of N2 is still very difficult due to high energy barrier for N≡N bond cleavage (~941 kJ/mol), large energy gap (~10.82 eV), and the lowest unoccupied molecular orbital [5]. The synthesis of NH3 by N2 hydrogenation is a complex reduction process with multiple reactions [6-8]. Industrially, the NH3 production from N2 and H2 was performed at high temperature (400−600 ℃) and pressure (20−40 MPa) by the well-known Haber-Bosch process [9-12], consuming enormous energy and concurrently releasing a large amount of CO2. Therefore, development of environmentally friendly NH3 synthesis techniques at ambient conditions is highly desirable.
Electrocatalytic N2 reduction reaction (NRR) at ambient conditions has been regarded as a promising NH3 synthesis method for replacing the traditional energy- and capital-intensive Haber-Bosch process [9-12]. To date, varieties of electrocatalysts have been synthesized and applied in electrocatalytic NRR to synthesize NH3, such as noble metal catalysts [13, 14], non-precious metal catalysts [15, 16] and metal-free carbon catalysts [17-19], demonstrating high NRR activities. Among non-precious metal catalysts, transition metal oxides have been proven to be efficient catalysts toward electrocatalytic NRR to NH3, confirming the oxygen vacancies (Vo) as the active centers [20-24]. As we know, niobium oxides have become important materials for catalysis applications [25] because of their superior catalytic activity and high physical/chemical stability. Recently, Kong et al. [26] reported Nb2O5 nanowires array as high-performance NRR electrocatalyst, demonstrating an NH3 yield rate of 1.58×10−10 mol s-1 cm-2 with a Faradaic efficiency (FE) of 2.26%. In their work, this good NRR activity can be ascribed to the important role of (001) facet of Nb2O5. The Nb2O5 nanofiber with high conductivity was also synthesized by Han and co-workers for electrocatalytic NRR to NH3 [27], showing an NH3 yield rate of 43.6 μg h-1 mgcat.-1 with an FE of 9.26%. In their study, this high NRR activity is mainly resulted from Nb-edge atoms of Nb2O5 (181) surface to polarize and activate N2 molecules. It is well known that pseudo hexagonal phase Nb2O5 has been confirmed to contain rich Vo [28], which could be a promising candidate as the electrocatalyst for NRR to NH3. In particular, pseudo hexagonal phase Nb2O5 nanostructured arrays (e.g., nanochannel array) could provide more excellent electron transfer pathways with exposed Vo active sites for high-efficiency NRR [29, 30].
Herein, highly ordered Nb2O5 nanochannel film (Nb2O5-NCF) was fabricated on niobium foil substrate by a facile anodization method. After thermal treatment in air, the as-fabricated Nb2O5-NCF with pseudo hexagonal phase contains rich oxygen vacancy defects, as the free-standing electrode, displaying high electrocatalytic NRR activity with an NH3 yield rate of 2.52×10−10 mol cm-2 s-1 and an FE of 9.81% at −0.4 V (vs. RHE) in 0.1 mol/L Na2SO4 solution (pH 3.2). During electrocatalytic NRR, it was found that the crystalline phase transformation of Nb2O5-NCF from pseudo hexagonal phase to hexagonal phase resulted in the electrochromism (EC) phenomenon and the decrease of NRR activity. This is mainly due to the decrease of Vo in hexagonal phase Nb2O5. The used Nb2O5-NCF electrode can be readily regenerated by low-temperature thermal treatment or applying an anodic potential, regaining high NH3 yield rate and FE with superior recycling reproducibility.
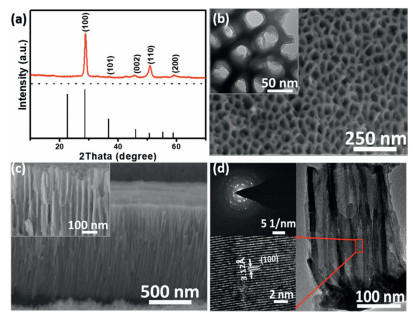
After anodization, the surface of niobium (Nb) foil showed dark brown and the surface colour of Nb foil became white after further treating in air at 450 ℃ (Fig. S1 in Supporting information). The XRD patterns (Fig. 1a) of the thermally treated sample exhibit a pseudo hexagonal phase (TT-phase) Nb2O5 (JCPDS No. 28-0317) [28]. The SEM images (Figs. 1b and c) show that the surface of the sample possesses porous structure, and a highly ordered nanochannel film with a thickness of ~2.0 μm forms on the Nb foil substrate (denoted as Nb2O5-NCF). The TEM image and selected area electron diffraction (SAED) patterns of the sample (Fig. 1d) confirm that the nanochannel walls are polycrystalline structure and the lattice fringes with interplanar distance of 3.12 Å can be ascribed to the (100) plane of TT-phase Nb2O5. The surface survey XPS spectrum of Nb2O5-NCF sample was provided in Fig. S2a (Supporting information), exhibiting Nb and O elements. The high-resolution Nb 3d XPS spectrum (Fig. S2b in Supporting information) indicates that the Nb 3d peaks at 209.6 and 206.9 eV can be assigned to Nb2O5 [31, 32].

|
Download:
|
| Fig. 1. (a) XRD pattern of Nb2O5-NCF. (b) Surface and (c) cross-sectional SEM images of Nb2O5-NCF. (d) TEM image and selected area electron diffraction (SAED) patterns for Nb2O5-NCF. | |
{kind=link}
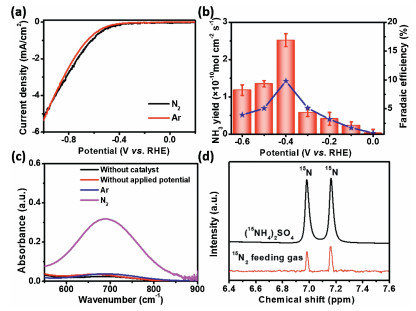
In this work, the fabricated Nb2O5-NCF was directly used as the cathodic electrode for NRR evaluation in 0.1 mol/L Na2SO4 solution (pH 3.2) using an H-type electrochemical cell. The linear sweep voltammograms (LSV) results (Fig. 2a) show that the cathodic current density of Nb2O5-NCF electrode in N2-saturated electrolyte is obviously larger than that obtained in Ar-saturated electrolyte when increasing applied potential from −0.3 V to −0.9 V, confirming good NRR activity of Nb2O5-NCF. Next, we investigated the influence of applied potential on the NRR performance of Nb2O5-NCF. During electrocatalytic NRR, the produced NH3 and/or N2H4 were determined by the indophenol blue [33] and Watt/Chrisp method [34], respectively. In this work, only NH3 product can be detected (Fig. S3 in Supporting information) while N2H4 is undetectable (Fig. S4 in Supporting information). Fig. 2b shows the NH3 yield rate and Faradaic efficiency (FE) of Nb2O5-NCF at different potentials, and corresponding chronoamperometric curves at each potential are shown in Fig. S5 (Supporting information). According to the UV–vis absorption spectra analysis (Fig. S3a) of NRR samples, the largest NH3 yield rate can reach 2.52×10−10 mol cm-2 s-1 with an FE of 9.81% at −0.4 V (vs. RHE) with the NRR time of 60 min (Fig. 2b). When the applied potential is more negative than −0.4 V (vs. RHE), the NRR performance of Nb2O5-NCF obviously decreases. This is mainly due to the competitive hydrogen evolution reaction (HER) concurrently happened on Nb2O5-NCF at more negative potentials [27]. To eliminate the environmental interference on the NH3 yielded during NRR, several control experiments were performed. The results (Fig. 2c) show that no NH3 product can be detected in 0.1 mol/L Na2SO4 solution without catalyst, with Nb2O5-NCF but without applied potential, or Ar-saturated electrolyte with Nb2O5-NCF at −0.4 V (vs. RHE), indicating no environmental interference on the produced NH3 by NRR process. In contrast, the UV–vis absorption spectra show stronger absorption peak at ~695 nm ascribed to the yielded NH3 determined by the indophenol blue method. To further confirm this, the isotopic labelling experiments using 15N2 as the feeding gas was also conducted. The 1H nuclear magnetic resonance (NMR) spectra (Fig. 2d) reveal that the chemical shift of doublet coupling can be observed for the NRR sample, due to15N in 15NH4+, suggesting the formed NH3 indeed originated from the Nb2O5-NCF catalyzed NRR process.

|
Download:
|
| Fig. 2. (a) LSV curves under Ar- or N2-saturated 0.1 mol/L Na2SO4 solution. (b) NH3 yield rate and FE of Nb2O5-NCF at different potentials. (c) UV–vis absorption spectra using the indophenol blue determination method under different conditions. (d) 1H NMR spectra for 15NH4+ in electrolyte using 15N2 as the feeding gas. | |
{kind=link}
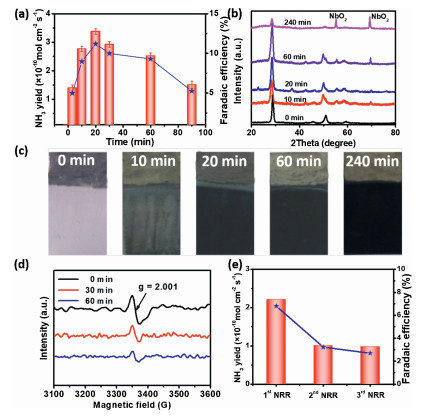
Subsequently, we evaluated the durability of Nb2O5-NCF electrode for NRR. It was found that the yielded NH3 amount during NRR initially rapidly increased. After 20 min, the increase trend of NH3 yield became slow (Fig. S6 in Supporting information). This results in an initial increase then decrease of NH3 yield rate, and the largest NH3 yield rate was obtained to be 3.38×10−10 mol cm-2 s-1 with an FE of 11.2% at -0.4 V (vs. RHE) (Fig. 3a) when NRR time of 20 min. The above experimental results suggested that the NRR activity of Nb2O5-NCF decreased after 20 min of reaction, which deserves a further investigation. During electrocatalytic NRR, the Nb2O5 electrode was found to transform to NbO2 species from XRD results (Fig. 3b), and electrochromism (EC) phenomenon was also observed, namely, the electrode surface color changed from white to dark with NRR time (Fig. 3c). This is mainly attributed to H+ ions insertion into the interlayer spacing of TT-phase Nb2O5, resulting in the crystalline phase transformation of Nb2O5 from TT-phase to T-phase, concurrently accompanied by part of Nb5+ reduction to Nb4+, thus the generation of EC phenomenon [35]. Seen from the XRD results of Nb2O5-NCF electrode with NRR time (Fig. 3b), compared to the pristine Nb2O5-NCF, the (100) diffraction peaks of Nb2O5-NCF with different NRR times apparently shift toward low angel direction in the XRD patterns, suggesting H+ ions insertion into interlayer spacing of Nb2O5 [36]. As we know, TT-phase Nb2O5 possesses more oxygen vacancies (Vo) than T-phase Nb2O5 [28]. These oxygen vacancies have been proven to be the catalytic active sites for N2 adsorption, activation and hydrogenation [26, 27]. The N2 firstly adsorbed on Vo sites of N2O5, and Vo-induced bands in Nb2O5 concurrently provided defect states for trapping excited electrons. Then these electrons were transferred and exchanged to the empty antibonding orbitals (p*) of the adsorbed N2. These adsorbed N2 (NN*) would be severely weakened and exhibited high activation due to the electron injection. It is conducive to hydrogenation for the N≡N bond to NH3. During NRR, the crystalline phase transformation from TT-phase to T-phase leads to the decrease of Vo concentration in Nb2O5, thus the decreased NRR performance.

|
Download:
|
| Fig. 3. (a) Time-dependence NH3 yield rate and FE during continuous NRR. (b) XRD patterns at different times during NRR. (c) The color change of Nb2O5-NCF with different reaction times during NRR. (d) EPR spectroscopy of Vo under different NRR times. (e) Recycling experiments using the used Nb2O5-NCF electrode with fresh 0.1 mol/L Na2SO4 electrolyte after 20 min of NRR for each cycle. | |
{kind=link}
Fig. 3d shows the electron paramagnetic resonance (EPR) spectroscopy of Nb2O5-NCF with different NRR times. In comparison with the pristine Nb2O5-NCF, the Vo concentration in Nb2O5 obviously decreased with the NRR time, primarily owing to H+ ions occupying the Vo sites thus resulting in the reduced active sites for N2 adsorption and activation.
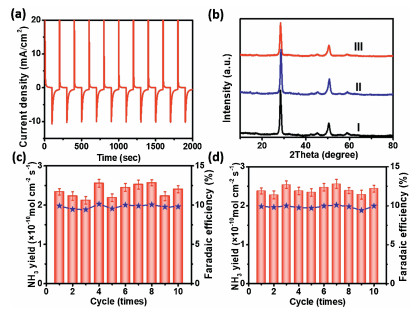
After 20 min of NRR, we replaced fresh 0.1 mol/L Na2SO4 electrolyte and conducted the recycling experiments using the used Nb2O5-NCF electrode and obtained obviously decreased NRR performance (Fig. 3e, each recycling experiment of 20 min). The above results further confirmed that with NRR time, the formation of T-phase Nb2O5 results in the decrease of Vo concentration, thus the reduced NRR performance. How to regenerate the electrode with high NRR activity is therefore critically important. We carried out the cyclic voltammetry (CV) experiments in 0.1 mol/L Na2SO4 electrolyte in the potential range from −1.2 V to 1.2 V (vs. RHE). The results (Fig. S7 in Supporting information) demonstrated that the oxidation peak lies at ~ −0.5 V (vs. RHE), corresponding to the extraction of H+ ions from the interlayer spacing of Nb2O5. The cathodic current density has a slight increase with the cycling number for Nb2O5-NCF and almost unchanged until 25th cycle, exhibiting an increased capacity for H+ ions insertion. The chronoamperometric measurements were performed by applying potentials of 0.6 and −0.6 V (vs. RHE) for 100 s. The comparison between the anodic and cathodic current intensities indicates a faster kinetic extraction than the insertion process, exhibiting an approximate bleaching time of 15 s while the colored time of 20 s (Fig. 4a). The reversible EC results mean that the used Nb2O5-NCF for NRR may be regenerated to recover its high NRR activity. For this, we tried to apply an anodic potential to realize this. As expected, when an anodic potential of 1.0 V (vs. RHE) was applied to the used Nb2O5-NCF electrode for 5 min, the T-phase Nb2O5 with part of Nb4+ can be readily transformed to TT-phase Nb2O5 (curve Ⅱ in Fig. 4b). This means that high NRR performance may be recovered by using the TT-phase Nb2O5 with more Vo sites. Utilizing such regeneration approach, the Nb2O5-NCF electrode displays superior recycling reproducibility with high NH3 yield rate and current efficiency (Fig. 4c).

|
Download:
|
| Fig. 4. (a) Chronoamperometric (CA) curves of Nb2O5-NCF for electrochromism (EC). (b) XRD patterns of Nb2O5-NCF before (curve Ⅰ) and after NRR with anodizing activation (curve Ⅱ) and low temperature annealing activation (curve Ⅲ). Recycling test of NRR for Nb2O5-NCF electrode by (c) anodizing activation and (d) low temperature annealing activation. | |
{kind=link}
Furthermore, we found that low-temperature treatment approach (60 ℃ for 0.5 h) is also feasible for regeneration of the used Nb2O5-NCF electrode for high-performance NRR (curve Ⅲ in Fig. 4b). The recycling reproducibility with high NH3 yield rate and current efficiency was shown in Fig. 4d. The EPR measurements of Nb2O5-NCF sample after 120 min NRR and subsequently low temperature annealing results also confirm this (Fig. S8 in Supporting information).
In summary, highly ordered Nb2O5 nanochannel film (Nb2O5-NCF) with rich Vo was fabricated on niobium foil substrate by a facile anodization method. As a free-standing electrode for NRR, the Nb2O5-NCF demonstrated high electrocatalytic activity toward NH3 synthesis. However, the NRR activity of Nb2O5-NCF decreased when the reaction time was over 20 min, due to a crystalline phase transformation of Nb2O5 from TT-phase to T-phase, resulting in the decrease of Vo concentration in Nb2O5, thus the reduced active sites for N2 adsorption and activation. However, the used Nb2O5-NCF with decreased NRR activity could be regenerated by applying an anodic potential or low-temperature thermal treatment approach, indicating superior recycling reproducibility with high NH3 yield rate and current efficiency. This work provides an efficient solution to the inactivation issue of an electrocatalyst for high-performance NRR to NH3.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work is financially supported by the National Key Research and Development Program of China (No. 2017YFA0207203) and the Natural Science Foundation of China (No. 51872292).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.020.
| [1] |
V. Smil, Nature 400 (1999) 415-415. DOI:10.1038/22672 |
| [2] |
V. Rosca, M. Duca, M.T. de Groot, M.T.M. Koper, Chem. Rev. 109 (2009) 2209-2244. DOI:10.1021/cr8003696 |
| [3] |
C.J. Pickett, J. Talarmin, Nature 317 (1985) 652-653. DOI:10.1038/317652a0 |
| [4] |
R. Zhang, Y. Zhang, X. Ren, et al., ACS Sustain. Chem. Eng. 6 (2018) 9545-9549. DOI:10.1021/acssuschemeng.8b01261 |
| [5] |
H. Jia, E.A. Quadrelli, Chem. Soc. Rev. 43 (2014) 547-564. DOI:10.1039/C3CS60206K |
| [6] |
M. Kitano, Y. Inoue, Y. Yamazaki, et al., Nat. Chem. 4 (2012) 934-940. DOI:10.1038/nchem.1476 |
| [7] |
K.A. Brown, D.F. Harris, M.B. Wilker, et al., Science 352 (2016) 448-450. DOI:10.1126/science.aaf2091 |
| [8] |
T. Oshikiri, K. Ueno, H. Misawa, Angew. Chem. Int. Ed. 55 (2016) 3942-3946. DOI:10.1002/anie.201511189 |
| [9] |
G.N. Schrauzer, T.D. Guth, J. Am. Chem. Soc. 99 (1977) 7189-7193. DOI:10.1021/ja00464a015 |
| [10] |
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) 1-14. |
| [11] |
L. Wang, M. Xia, H. Wang, et al., Joule 2 (2018) 1055-1074. DOI:10.1016/j.joule.2018.04.017 |
| [12] |
T.L. Frolicher, E.M. Fischer, N. Gruber, Nature 560 (2018) 360-364. DOI:10.1038/s41586-018-0383-9 |
| [13] |
Y. Yao, S. Zhu, H. Wang, H. Li, M. Shao, J. Am. Chem. Soc. 140 (2018) 1496-1501. DOI:10.1021/jacs.7b12101 |
| [14] |
H.K. Lee, C.S.L. Koh, Y.H. Lee, et al., Sci. Adv. 4 (2018) eaar3208(1-8). |
| [15] |
J. Kong, A. Lim, C. Yoon, et al., ACS Sustain. Chem. Eng. 5 (2017) 10986-10995. DOI:10.1021/acssuschemeng.7b02890 |
| [16] |
L. Zhang, X. Ji, X. Ren, et al., Adv. Mater. 30 (2018) 1800191. DOI:10.1002/adma.201800191 |
| [17] |
S. Chen, S. Perathoner, C. Ampelli, et al., Angew. Chem. Int. Ed. 56 (2017) 2699-2703. DOI:10.1002/anie.201609533 |
| [18] |
S. Mukherjee, D.A. Cullen, S. Karakalos, et al., Nano Energy 48 (2018) 217-226. DOI:10.1016/j.nanoen.2018.03.059 |
| [19] |
W. Li, T. Wu, S. Zhang, et al., Chem. Commun. 54 (2018) 11188-11191. DOI:10.1039/c8cc06000b |
| [20] |
L. Huang, J. Wu, P. Han, et al., Small Methods 3 (2019) 1800386. DOI:10.1002/smtd.201800386 |
| [21] |
R. Manjunatha, A. Karajić, V. Goldstein, A. Schechter, ACS Appl. Mater. Interfaces 11 (2019) 7981-7989. DOI:10.1021/acsami.8b20692 |
| [22] |
J. Han, X. Ji, X. Ren, et al., J. Mater. Chem. A 6 (2018) 12974-12977. DOI:10.1039/C8TA03974G |
| [23] |
C. Fang, T. Bi, X. Xu, et al., Adv. Mater. Interfaces 6 (2019) 1901034. DOI:10.1002/admi.201901034 |
| [24] |
W. Fu, P. Zhuang, M.O. Chee, et al., ACS Sustain. Chem. Eng. 7 (2019) 9622-9628. DOI:10.1021/acssuschemeng.9b01178 |
| [25] |
I. Nowak, M. Ziolek, Chem. Rev. 99 (1999) 3603-3624. DOI:10.1021/cr9800208 |
| [26] |
W. Kong, Z. Liu, J. Han, et al., Inorg. Chem. Front. 6 (2019) 423-427. DOI:10.1039/c8qi01049h |
| [27] |
J. Han, Z. Liu, Y. Ma, et al., Nano Energy 52 (2018) 264-270. DOI:10.1016/j.nanoen.2018.07.045 |
| [28] |
Y. Huang, Y. Zhang, X. Hu, Sol. Energy Mater. Sol. C 77 (2003) 155-162. DOI:10.1016/S0927-0248(02)00318-5 |
| [29] |
H. Cui, G. Zhu, Y. Xie, et al., J. Mater. Chem. A 3 (2015) 11830-11837. DOI:10.1039/C5TA01544H |
| [30] |
H. Yu, L. Xu, H. Wang, et al., Electrochim. Acta 295 (2019) 829-834. DOI:10.3390/e21090829 |
| [31] |
M.K. Bahl, J. Phys. Chem. Solids 36 (1975) 485-491. DOI:10.1016/0022-3697(75)90132-8 |
| [32] |
T. Joshi, T.R. Senty, P. Borisov, A.D. Bristow, D. Lederman, J. Phys. D Appl. Phys. 48 (2015) 335308. DOI:10.1088/0022-3727/48/33/335308 |
| [33] |
P.L. Searle, Analyst 109 (1984) 549-568. DOI:10.1039/an9840900549 |
| [34] |
D. Bao, Q. Zhang, F. Meng, et al., Adv. Mater. 29 (2017) 1604799. DOI:10.1002/adma.201604799 |
| [35] |
C. Shi, K. Xiang, Y. Zhu, et al., Electrochim. Acta 246 (2017) 1088-1096. DOI:10.1016/j.electacta.2017.06.109 |
| [36] |
R. Romero, E.A. Dalchiele, F. Martín, D. Leinen, J.R. Ramos-Barrado, Sol. Energy Mater. Sol. C 93 (2009) 222-229. DOI:10.1016/j.solmat.2008.10.012 |