 2021, Vol. 32
2021, Vol. 32 


b School of Chemical Engineering, Sichuan University, Chengdu 610065, China;
c Department of Water Management, Section Sanitary Engineering, Delft University of Technology, PO Box 5048, 2600 GA, Delft, the Netherlands
Electrochemical advanced oxidation process (EAOP) is an intriguing technology in water treatment due to it can efficient produce hydroxyl radicals ·OH, which is a reactive species could decompose refractory organics with a high second order reaction kinetics in 106–1010 L mol−1 s−1 order [1-3]. As an vital component in EAOP, the choice of anode is significant for environmental applications related to wastewater treatment using "nonactive" anode [1-3], which owns high overpotential toward oxygen evolution reaction and could generate·OH with "quasi-free" state [1, 4-6]. And then, the organic pollutants undergoing the process of the reaction involved with·OH (Eqs. 1 and 2) [6-9]. Under this context, "active" anode produces oxygen favorable Eq. 3, which enhance the oxygen evolution reaction (OER) and the partial oxidation of the contaminants. While "nonactive" anode favors the mineralization of the organic compounds through an indirect mechanism involving a strong oxidizing mediator (·OH radicals) [6-8].

|
(1) |

|
(2) |
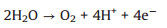
|
(3) |
Thus, a precondition for an ideal EAOP anode should maintain a high oxygen evolution potential (OEP) to avoid side OER reaction, and a weak adsorption energy towards ·OH to produce "quasi-free" ·OH.
The anodes with high overpotentials for oxygen evolution, such as β-PbO2, Ti/SnO2, and boron-doped diamond (BDD), are intensively investigated and employed in the EAOP [10-12]. Concerning the typical "nonactivity" characteristic, the BDD is the state-of-the-art EAOP catalyst, since it can produce quantity of "quasi-free" ·OH [4]. Learning from previous studies, the "nonactive" characteristic of BDD anode was attributed to the surface carbon configuration, the sp2 and sp3 carbon on BDD surface [13]. Nevertheless, overcoming its inherent high cost and manageability remain as main challenges to achieve a pilot-scale operation [13], thus the development of other carbonaceous EAOP anode is highly desirable.
Graphdiyne (GDY), since found by Li's group, which could be obtained by a facile cross-coupling reaction on copper foil using hexaethynylbenzene as the precursor [14, 15]. GDY electrode material is regarded as an emerging "star" carbonaceous material in lithium-ion battery anodes, hydrogen evolution reaction (HER), and oxygen reduction reaction (ORR) due to its unique 2-dimension (2D) molecular configuration of sp and sp2 carbon [15, 16]. Inspired by the surface carbon hybridization configuration of BDD, it is anticipated that GDY is another alternative EAOP anode for producing abundant hydroxyl radicals. Therefore, the theoretical predictive work to evaluate GDY as a desired EAOP anode is highly appreciated.
Herein, it is for the first time to build the correlation of OER with ·OH generation based on first-principle calculations by taking GDY as a model catalyst. The electrocatalytic behavior of "nonactive" (GDY) anode towards the OER and enhanced activity towards ·OH generation were investigated through density functional theory (DFT) method. All the results indicated the GDY is an ideal "nonactive" anode, which can produce aqueous ·OH ("free" hydroxyl radicals) rather than surface-bound ·OH to construct an efficient EAOP for pollutant removal.
In this work, all calculations were performed using the Vienna ab initio simulation package (VASP) code [16]. Projector augmented wave (PAW) was used to describe the interactions of the core electrons [17], in which the exchange-correlation interactions were described by the generalized gradient approximation (GGA) in the form of the Perdew–Burke–Ernzerhof (PBE) functional [18]. The cut-off energy was set as 500 eV, and the convergence tolerance for residual force and energy on each atom during structure relaxation were 0.01 eV/ Å and 10−5 eV, respectively. The van der Waals interaction was taken into account using the semi-empirical dispersion-corrected DFT-D2 scheme proposed by Grimme [19]. The vacuum space was 15 Å to avoid the interaction of periodic images. The Brillouin zone was sampled with Monkhorst-Pack 5×5 × 1 k-points.Ab initio molecular dynamics (AIMD) simulations were also taken into consideration to check the dynamical stability of GDY, and the algorithm of the Nose thermostat was used to simulate a canonical ensemble [20].
As reported [21], at acidic condition, the OER mechanism follows four-stage pathway that can be summarized as follows (Eqs. 4-7):

|
(4) |
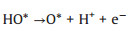
|
(5) |
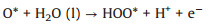
|
(6) |
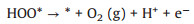
|
(7) |
where (l) and (g) refer to the liquid and gas phase, respectively. * is the adsorptive site on the GDY, and O*, HO* and HOO* are adsorbed intermediates, and more importantly, the HO* is not hydroxyl radical (·OH).
For each step [22], ΔG can be defined by the following Eq. 8 [23].

|
(8) |
The adsorption free energy (Eads) of adsorbates is defined by Eq. 9 [23].

|
(9) |
The overpotentials of the OER (ηOER) for GDY can be obtained by Eq. 10 [22, 23].
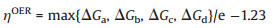
|
(10) |
where ΔGa, ΔGb, ΔGc, ΔGd is Gibbs free energy of reactions in Eqs. 4-7.
Fig. 1 shows the optimized configuration of the pure 2×2 × 1 GDY supercell and density of state (DOS). GDY exhibits a planar sheet-like configuration with the formation of 6C-hexagon and 18C-hexagon. The bond length of C-C in 6C-hexagon is 1.43 Å, and the bond length of C-C and C≡C in the diacetylenic links of the 18C-hexagon is 1.39 and 1.23 Å, respectively [24]. The calculated band gap of pure GDY is 0.38 eV, agreeing well with previous theoretical results [25]. This indicates that the GDY possess a comparable electric conductivity than that commonly used EAOP anodes such as β-PbO2 and SnO2. As prior mentioned, the overpotential of OER is a pivotal descriptor to describe the EAOP activity of an anode catalyst [21], thus we calculated the reaction free energy of the adsorbed species HO*, O*, and HOO* adsorbed on GDY.

|
Download:
|
| Fig. 1. (a) The top view of the optimized structure of pure graphdiyne (GDY). (b) The density of states (DOS) for the pure GDY. The Fermi level is set at zero with the black line. | |
{kind=link}
Fig. 2 exhibits the calculated free energy diagram for pure GDY. It can be seen that the HO* formation step is the rate-limiting step with the high energy barrier of 3.18 eV. This implies that the overpotential for the OER (ηOER), occurring on the pure GDY, is 1.95 V. This overpotential parameter is comparable with BDD anode (~1.6 V) [4]. The high overpotential of GDY anode indicates that OER is not favorable during the EAOP process, therefore GDY is beneficial to produce·OH for organic oxidation reaction. Besides, the water oxidation reaction on anode is mediated by the four electrons processes. For OER process, the 1e− intermediate is HO*, 2e− intermediate is O*, and 4e− product is O2. For EAOP process, ·OH is the 1e− product, H2O2 is the 2e− product and O2 is the ultimate product. In this context, the first step in OER is crucial for hydroxyl radical generation, i.e., the 1e− intermediate HO* production reaction of OER is the competitive reaction for ·OH production of EAOP. Based on Nørskov's study, the thermodynamic equilibrium potential of hydrated hydroxyl radicals (1 ppm ·OH with room temperature, 298 K) is 2.4 eV [21]. In this current research, the calculated thermodynamic potential of HO* on GDY anode is 3.18 eV, which is 0.78 eV larger than that of·OH. This indicates that the water oxidation of ·OH is thermodynamically favorable on GDY anode, whereas the oxidation of water will be more inclined to produce ·OH rather than O2. On the other hand, BDD anode is well-known contains loosely adsorbed ·OH [26], resulting in higher decomposition of the organic compounds. As shown in Table S1 (Supporting information), the adsorption free energy of HO* on GDY is 2.85 eV, which indicates that the HO* is adsorption unfavorable process. Thus once HO* is formed, it is equally favorable to desorb and form free·OH (aq). Besides, as a widely used EAOP anode, the water oxidation pathway on boron-doped diamond anode was also investigated. As shown in Table S2 (Supporting information), the calculated thermodynamic potential of HO* on commercial BDD anode is 2.82 eV, thus the BDD anode owns a theoretical OER overpotential of 1.59 V. Furthermore, in order to give a more clear demonstration, the linear sweep voltammetry (LSV) polarization curve also recorded. As shown in Fig. S1 (Supporting information), in 0.05 mol/L H2SO4 solution, the BDD anode exhibit poor oxygen evolution performance. The ηOER is 1.6 V at the current density of 10 mA/cm2, which in line with well with the obtained result of DFT calculation (1.59 V). As a comparison, the thermodynamic potential of HO* on GDY anode is 3.18 eV, a value of 0.36 V higher than that of BDD, this evidently demonstrates that the GDY anode is a typical "nonactive" anode which can produce quantity of "free" for decomposing organic pollutants in bulk or liquid/anode interface effectively.

|
Download:
|
| Fig. 2. The free energy diagram for the OER at zero potential over pure GDY. The red line is the rate-determining step for OER. | |
{kind=link}
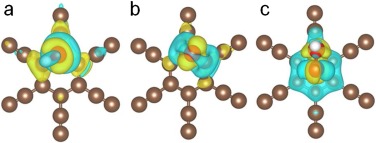
To understand the water discharge on GDY anode systemically, the overall OER process on GDY was investigated. The OER involves three conversion steps, from the adsorbed HO* to O*, and then transforming to HOO*, as shown in Fig. S2 (Supporting information). As discussed above, the adsorption of HO* with GDY is not thermodynamic feasible, so it is adverse for the generation of O* and HOO*. Therefore, the water oxidation reaction path on GDY anode is favorable for producing ·OH rather than O2. To gain deeper insight into activity towards high OEP of GDY, the Bader charge, charge density difference and partial density of states (PDOS) of oxygen-containing intermediates adsorbed on the pure GDY are systematically analyzed. By analyzing the calculated Bader charge, the adsorbed oxygen-containing intermediates (HO*, O* and HOO*) on pure GDY are all negatively charged by 0.28, 0.79, and 0.14 e, respectively, indicating that the charge transfer from pure GDY to intermediates. As exhibited by the charge density difference (Fig. 3), it can be seen that the accumulation regions are mainly around the adsorbed oxygenated species.

|
Download:
|
| Fig. 3. The charge density difference of (a) HO* adsorbed on the pure GDY, (b) O* adsorbed on the pure GDY, and (c) HOO* adsorbed on the pure GDY. The brown, red and white balls stand for carbon, oxygen and hydrogen atoms, respectively. The yellow areas mean that the accumulation of electrons and the blue region exhibits the depletion of electrons (isovalue of 0.002 e/Å3). | |
{kind=link}
The above results can be further explicated by the PDOS analyses. The PDOS curves are illustrated in Fig. S3 (Supporting information). A hybridizations between the C-p states and O-p states were observed. Moreover, the hybridization between O-p states and C-p of O* adsorbed pure GDY is larger than that of HO* and HOO* on pure GDY, resulting in the stronger interaction strength than that of other two species on pure GDY This observation is in agreement with the above results of Bader charge and charge density difference analysis.
The anode stability is an important parameter responsible for the anode service lifetime towards the application of EAOP technology. In order to investigate the stability of GDY anode, the AIMD simulations were carried out to examine its dynamical stability under 300 K for 10 ps with a time step of 2 fs. The GDY monolayer undergoes a minimum change, but neither significant chemical bond rupture nor remarkable structural reconstruction were observed at the end of the MD simulations as shown in Fig. 4. The energies and temperatures oscillate near the equilibrium state, indicating the good thermodynamic stability of the GDY, and possible long service lifetime in practical application.

|
Download:
|
| Fig. 4. Total energy and temperature evolutions of the GDY as function of time for AIMD are plotted, and insert the snapshot of the GDY structure at the end of MD simulation. | |
{kind=link}
The control of the electrocatalysis of the H2O oxidation is prerequisite since depending on the properties of the catalysts, it can enhance and retain ·OH yield ("nonactive" anode for OER), or continuing to evolve O2 ("active" anode for OER). In this study, GDY anode was found a large OER overpotential (ηOER=1.95 V) and a weak sorptive ability towards oxygen evolution intermediates (HO*). The free energy of adsorbed HO* was taken as a descriptor to evaluate ·OH and O2 yield in thermodynamic viewpoint for GDY catalyst. The high Gibbs energy change of HO* (3.18 eV) on GDY anode makes the selective production of ·OH (ΔG=2.4 eV) is thermodynamically favorable. Besides, the Ab initio molecular dynamics (AIMD) simulation indicate the GDY anode is with good thermodynamic stability, which meets the requirement of engineering stability. Overall, our research proposes GDY can be regarded as an alternative "nonactive" anode for effective degradation refractory organic compounds in electrochemical oxidation (EO) process, and DFT may be a general approach which will be widely employed in environmental electrochemistry.
Declaration of competing interestThe author declare that they have no financial and personal relationships with other people or organizations that can inappropriately influence their work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled "Graphdiyne as a Novel Nonactive Anode for Wastewater Treatment: A Theoretical Study".
AcknowledgmentsWe thank the support from the National Key Research and Development Program of China (No. 2017YFE9133400), Pre-research Fund of Jiangsu Collaborative Innovation Center of Technology and Material of Water Treatment (No. XTCXSZ2020-3).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.017.
| [1] |
J. Li, Y. Li, Z. Xiong, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 2139-2146. DOI:10.1016/j.cclet.2019.04.057 |
| [2] |
J.F. Zhong, D.L. He, Z. Zhou, Y.B. Xu, Chin. Chem. Lett. 19 (2008) 319-323. DOI:10.1016/j.cclet.2007.12.017 |
| [3] |
R.W. Yan, B.K. Jin, Chin. Chem. Lett. 24 (2013) 159-162. DOI:10.1016/j.cclet.2013.01.023 |
| [4] |
C.A. Martinez-Huitle, S. Ferro, Chem. Soc. Rev. 35 (2006) 1324-1340. DOI:10.1039/B517632H |
| [5] |
C.A. Martínez-Huitle, M.A. Rodrigo, I. Sires, O. Scialdone, Chem. Rev. 115 (2015) 13362-13407. DOI:10.1021/acs.chemrev.5b00361 |
| [6] |
B. Marselli, J. Garcia-Gomez, P.A. Michaud, M. Rodrigo, C. Comninellis, J. Electrochem. Soc. 150 (2003) D79-D83. DOI:10.1149/1.1553790 |
| [7] |
B.P. Chaplin, Environ. Sci.-Proc. Imp. 16 (2014) 1182-1203. DOI:10.1039/C3EM00679D |
| [8] |
D. Bejan, E. Guinea, N.J. Bunce, Electrochim. Acta 69 (2012) 275-281. DOI:10.1016/j.electacta.2012.02.097 |
| [9] |
D. García-Osorio, R. Jaimes, J. Vazquez-Arenas, R. Lara, J. Alvarez-Ramirez, J. Electrochem. Soc. 164 (2017) E3321-E3328. DOI:10.1149/2.0321711jes |
| [10] |
J.M. Aquino, R.C. Rocha-Filho, L.A. Ruotolo, N. Bocchi, S.R. Biaggio, Chem. Eng. J. 251 (2014) 138-145. DOI:10.1016/j.cej.2014.04.032 |
| [11] |
A. Polcaro, S. Palmas, F. Renoldi, M. Mascia, J. Appl. Electrochem. 29 (1999) 147-151. DOI:10.1023/A:1003411906212 |
| [12] |
V. Schmalz, T. Dittmar, D. Haaken, E. Worch, Water Res. 43 (2009) 5260-5266. DOI:10.1016/j.watres.2009.08.036 |
| [13] |
F. Souza, C. Saéz, M. Lanza, P. Cañizares, M. Rodrigo, Electrochim. Acta 187 (2016) 119-124. DOI:10.1016/j.electacta.2015.11.031 |
| [14] |
Y. Li, L. Xu, H. Liu, Y. Li, Chem. Soc. Rev. 43 (2014) 2572-2586. DOI:10.1039/c3cs60388a |
| [15] |
G. Li, Y. Li, H. Liu, et al., Chem. Comm. 46 (2010) 3256-3258. DOI:10.1039/b922733d |
| [16] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [17] |
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758-1775. DOI:10.1103/PhysRevB.59.1758 |
| [18] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [19] |
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799. DOI:10.1002/jcc.20495 |
| [20] |
G.J. Martyna, M.L. Klein, M. Tuckerman, J. Chem. Phys. 97 (1992) 2635-2643. DOI:10.1063/1.463940 |
| [21] |
J.K. Nørskov, J. Rossmeisl, A. Logadottir, et al., J. Phys. Chem. B 108 (2004) 17886-17892. DOI:10.1021/jp047349j |
| [22] |
P. Atkins, Paula J. Atkins' physical chemistry, Oxford University Press Oxford, U.K., 2006
|
| [23] |
A. Valdes, Z.W. Qu, G.J. Kroes, J. Rossmeisl, J.K. Nørskov, J. Phys. Chem. C 112 (2008) 9872-9879. DOI:10.1021/jp711929d |
| [24] |
P. Wu, P. Du, H. Zhang, C. Cai, Phys. Chem. Chem. Phys. 16 (2014) 5640-5648. DOI:10.1039/C3CP55121K |
| [25] |
A. Ivanovskii, Prog. Solid State Ch. 41 (2013) 1-19. DOI:10.1016/j.progsolidstchem.2012.12.001 |
| [26] |
A. Kapałka, G. Fóti, C. Comninellis, Electrochim. Acta 53 (2007) 1954-1961. DOI:10.1016/j.electacta.2007.08.066 |