 2021, Vol. 32
2021, Vol. 32 


b College of Mechanics and Materials, Hohai University, Nanjing 210098, China
Currently, air pollution is one of the most critical problems for human health and environment [1]. One of the main types of air pollutants is volatile organic compounds (VOCs), which comprises a variety of compounds, such as alkanes, alkenes, alkynes, aromatic and halogenated hydrocarbons [2, 3]. It is reported that long-term exposure to high concentrations of VOCs can cause serious carcinogenic, mutagenic, and teratogenic effects to human beings [4, 5]. In addition, VOCs are also the precursors for many other atmospheric pollutants, such as ozone, secondary aerosol, and chemical smog [6, 7]. Therefore, it is urgent to remove VOCs from atmospheric to protect human health and mitigate air pollution.
At present, the research on VOCs treatment has become one of the hottest topics in the field of environmental protection [8-10]. There are various technologies to remove VOCs, such as biodegradation [11], photocatalytic oxidation [12], combustion [13] as well as adsorption [14]. Among these technologies, adsorption is a simple and low-cost process, it has high efficiency and wide applications. The adsorbent plays a critical role in the wide use of adsorption. Various adsorbents for VOCs have already been reported, such as zeolites, activated carbon, metal organic frameworks (MOFs) [15, 16].
Recently, the low dimensional materials show the potential to remove pollutants [17]. Especially, with high specific surface area, the two-dimensional (2D) materials have very large adsorption capacity as adsorbents [18]. For example, by using first-principle calculations, Su et al. demonstrated that the pristine 2D materials show unsatisfactory capacity in adsorbing VOCs, while the doped 2D materials, such as graphene and C2N, are excellent adsorbents for some VOCs [19]. Liu et al. also found that the Al-decorated porous graphene shows outstanding selective adsorption capacity for carbonyl-containing volatile organic compounds [20]. However, the complex processes for modification limit the application of 2D adsorbents. Therefore, it is desirable to explore pristine and simple 2D materials as VOCs adsorbents. Borophene is a monolayer sheet with single element boron. The research of borophene has been going on for many years [21-26], although it was first synthesized in 2016 [27]. It was reported that borophenes withχ3 and β12 structures were synthetized on Ag(111) surface employing molecular beam epitaxy (MBE) by Feng et al. [27]. As the first synthesized borophenes in experiment, many characteristics of χ3 and β12 borophene are observed. The borophene show a certain antioxidant capacity, indicating that the structures can exist stably in natural environment. The metallicity of χ3 and β12 borophene also reveals the potential application in electronic device [28]. As a 2D material, borophene has high specific surface area, which is important for absorbents. Liu et al. have proved that some small gas molecules (CO, NO and NH3) can be adsorbed steady on borophene [29], which confirms the potential as an excellent gas adsorbent. Therefore, it is expected that borophene might be an adsorbent for VOCs.
Density functional theory (DFT), an important method to study the electron properties of object, is usually used to investigate and understand the nature of chemical behaviors such as photocatalysis [30] and adsorption [31]. Therefore, DFT are employed to investigate the adsorption properties of typical VOCs molecules onto χ3 and β12 borophene in this work. Some simple and typical VOCs molecules, including ethane (C2H6), ethylene (C2H4), formaldehyde (H2CO), methanol (H3COH), formic acid (HCOOH), methyl chloride (H 3CCl), benzene (C6H6) and toluene (Ph-CH3), are considered for their adsorption on borophene. The geometric structure and the electron properties are analyzed for the all adsorption configurations to reveal the interactions between borophenes and the VOCs. This work is found to be beneficial to discover new treatment and detection methods for VOCs.
All DFT calculations are performed by Dmol3 modulus in Materials Studio Package [32]. The calculations using the local density approximation (LDA) underestimate equilibrium distances and overestimate bond energy. Thus, the structure optimization and energy calculations of the most stable geometries in this study are based on the generalized-gradient approximation (GGA) function with the Perdew–Burke–Ernzerhof (PBE) correction [33]. The method proposed by Grimme for DFT-D correction is chosen to elucidate the weak van der Waals interaction [34]. The global orbital cutoff for all atoms is set to 4.5 Å. The DFT semi-core pseudopotentials (DSPP) core treatment is implemented for relativistic effects, which replaces core electrons by a single effective potential [35]. The k-point is set at 5×5 × 1 for all slabs, which brings out the convergence tolerance of energy of 1.0×10−6 hartree (1 hartree = 27.2114 eV), and that of maximum force of 0.002 hartree/ Å.
To understand the adsorption properties of VOCs on borophene, the adsorption energy of VOCs molecules adsorbed onto borophene Eads is calculated and defined as:
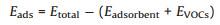
|
(1) |
where Etotal, Eadsorbent, and EVOCs are the total energies of the system with the gas molecule adsorbed on the pristine borophene, an isolated pristine borophene, and the VOCs molecule in the ground state, respectively.
In this study, χ3 and β12 borophenes are chosen to investigate the performance for adsorbing VOCs. The optimized structures of 3×4 × 1 χ3 borophene and 2×4 × 1 β12 borophene are shown in Figs. S1a and b (Supporting information) respectively, where the borophenes are constructed by repetitive hexagon holes and triangular boron sheet in a plat surface. The hexagonal triangular density is η=1/5 and η=1/6 for χ3 and β12 borophene, respectively [28, 36]. The optimized lattice parameters for the unit cell χ3 borophene are a = 4.47 Å, b = 2.90 Å and γ = 71.21°, which are in good agreement with previously reported results (a = 4.44 Å, b = 2.90 Å and γ =70.95°) [[ 28]. The unit cell constants of β12 borophene are a = 2.93 Å, b = 5.10 Å and γ =90°, similar as the cell parameters in the previous report of Wu et al., i.e., a = 2.92 Å, b = 5.05 Å andγ=90° [28].
Furthermore, the electronic structures of borophenes are also considered. The band structure of unit cell χ3 and β12 borophene are shown in Figs. S1c and d (Supporting information), respectively. In the figures, specific symmetry k-points and path in Brillouin zone for the band structure are tagged out. As show in Fig. S1c, there are many bands crossing the Fermi level along M-G, G-F, F-K, B-G and G-K, indicating the electrons in valence band easily shift to conduction band through these bands. Similarly, the bands crossing Fermi level in Fig. S1d mainly locate at the path G-X, S-Y, indicating the β12 borophene also has the capability of electron conduction. Interestingly, the metallicity of the 2D borophenes are different from the 3D α-rhombohedral boron B12, which is nonmetal with an indirect band gap [37].
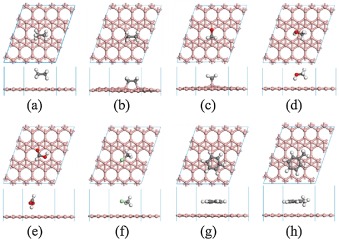
After understanding the atomic structure of χ3 and β12 borophenes, the adsorption of various typical VOCs molecules (C2H6, C2H4, H2CO, H3COH, HCOOH, H3CCl, C6H6 and Ph-CH3) on the two different borophenes is investigated. The most stable configurations of the VOCs molecules adsorbed on χ3 and β12 borophene are shown in Fig. 1 and Fig. S2 (Supporting information), respectively. Compared with the structures of pristine borophenes in Fig. S1, there is no distinct deformation of borophenes after adsorbing VOCs, except for the adsorption of C2H4 and H2CO molecules as shown in Figs. 1b, c and Figs. S2b, c, where chemical bonds between C2H4 or H2CO and borophene are formed. It is obvious that two B atoms of the borophene protrude from the plane due to the chemical adsorption of C2H4 and H2CO molecules. Moreover, the borophenes also induce structure deformation of the adsorbed C2H4 and H2CO molecules. For C2H4, the length of C=C bond extends from 1.33 Å to 1.55 Å on χ3 borophene and to 1.56 Å on β12 borophene, as listed in Table S1 (Supporting information), indicating the breaking of C=C bond into C-C bond. In addition, the angle formed by the two C-H bonds reduces to 106.513° on χ3 borophene and 106.403° on β12 borophene from the gas-phase value of 115.673°, indicating that the C atoms of the adsorbed C2H4 become sp3 hybrids under the interaction with borophenes. Then the C atoms bind with protruded B atoms to form two C-B bonds (bond length = 1.74 Å) on χ3 borophene and two C-B bonds (bond length = 1.68 Å) on β12 borophene. For H2CO, the C=O bond breaks into C-O bond with the bond length extending from 1.21 Å to 1.46 Å on χ3 borophene and to 1.45 Å on β12 borophene. Then the C and O atoms of H2CO bind with the protruded B atoms of borophenes to form a C-B bond (bond length = 1.65 Å) and an O-B bond (bond length = 1.42 Å) on χ3 borophene and a C-B bond (bond length = 1.64 Å) and an O-B bond (bond length = 1.40 Å) on β12 borophene (Table S1). For all the other adsorbed VOCs molecules investigated, the shortest distance between the VOCs molecules and borophenes can be defined by the distance between the bottom H atom in molecule and the B atom of borophene from the adsorption configuration in Fig. 1 and Fig. S2, and the corresponding results are listed in Table S2 (Supporting information). It can be found that the distance is in the range of 2.53 Å–3.22 Å, which is not short enough for the formation of chemical bond between VOCs and borophenes. Therefore, only weak interaction is present between these VOCs molecules and borophenes.

|
Download:
|
| Fig. 1. The most stable adsorption configurations of C2H6 (a), C2H4 (b), H2CO (c), H3COH (d), HCOOH (e), H3CCl (f), C6H6 (g) and Ph-CH3 (h) on χ3 borophene. | |
The adsorption energy is another important parameter for adsorption. As shown in Table 1, the adsorption energy for C2H4 adsorbed on χ3 and β12 borophenes is 1.14 eV and 0.80 eV respectively. For the adsorption of H2CO, the corresponding adsorption energy on χ3 and β12 borophene are 1.21 eV and 0.79 eV respectively, larger than that of H2CO on other pristine 2D materials, such as graphene (0.29 eV) [38] and MoS2 (0.11 eV) [39]. Therefore, the strong interaction between borophenes and the C2H4 and H2CO can be more confirmed. It should be noted that the adsorption energies for C6H6 and Ph-CH3 on χ3 and β12 borophene are between 0.60 eV to 0.77 eV, which is relatively large for physical adsorption. Considering the above analysis of geometric configuration and electron properties below, they still belong to physical adsorption and the large adsorption energy may result from the relatively greater molecular mass of the C6H6 and Ph-CH3 molecules. For the adsorption of all the other VOCs, the adsorption energy is between 0.23 eV to 0.37 eV, which are in the rage of physical adsorption. Combined with the large distance between boropehens and these VOCs molecule, it can be found that the Van der Waals force plays a main role during the adsorption.
|
|
Table 1 Adsorption energy and electron transfer number calculated by Mulliken analysis for different VOCs molecules adsorbed on borophenes. |
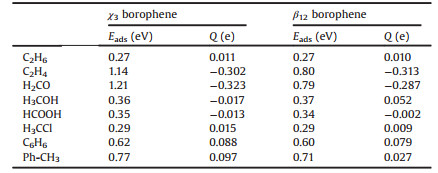
{kind=link}
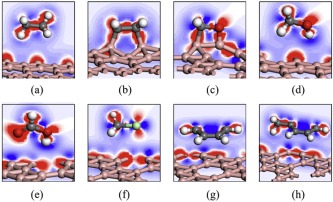
The analysis of geometric configurations reveals the strong interaction between borophenes and the C2H4 or H2CO with the formation of chemical bonds. To further confirm the formation of chemical bonds, the analysis of electron properties is necessary. The deformation of electronic densities for typical VOCs adsorbed on borophenes are calculated and shown in Fig. 2 and Fig. S3 (Supporting information). In these figures, the red regions represent the areas of electron accumulation, while the blue regions reflect the areas of electron loss. In Fig. 2b and Fig. S3b, the two new C-B bonds, formed by C2H4 adsorption on borophenes, are covered by red regions, indicating that the new bonds formation due to the overlap of electronic cloud. It can also be found that distinct red regions are on the C-B and O-B bond of borophenes with H2CO adsorption as shown in Fig. 2c and Fig. S3c. For all the other VOCs molecules adsorbed on the borophenes, however, there are no distinct regions existed between the molecules and adsorbents, confirming that only physical adsorptions existed. The electron density results agree with the above-mentioned conclusion of geometric configuration.

|
Download:
|
| Fig. 2. Deformation of electronic densities of C2H6 (a), C2H4 (b), H2CO (c), H3COH (d), HCOOH (e), H3CCl (f), C6H6 (g) and Ph-CH3 (h) on χ3 borophene. The red regions represent the areas of electron accumulation, while the blue regions reflect the areas of electron loss. | |
{kind=link}
Electron transfer (Q) in adsorption system is also a common analysis method to understand the adsorption. The electron transfer of the adsorbed molecules and borophenes can be calculated by Mulliken analysis and the results are listed in Table 1. It can be found that the electron transfers are between 0.010 e to 0.088 e in the system of physical adsorptions involving C2H6, H3COH, HCOOH, H3CCl, C6H6 and Ph-CH3. Among the physical adsorptions system, the electron transfers for C6H6 adsorbed on χ3 and β12 borophene are relatively large of 0.088 e and 0.079 e, similar as that of its physical adsorption on graphene (0.02 e) [38] and C2N (0.018 e) [19]. On the contrary, more electron transfer occurs in chemical adsorption of C2H4 and H2CO adsorbed on the borophenes. The electronic charges of about 0.302 e and 0.313 e are respectively transferred from χ3 and β12 borophene to the C2H4 molecule, which are almost one order larger than that in the above physical adsorptions. In addition, the H2CO molecule adsorbed on borophenes induces around 0.323 e and 0.287 e charge transfer from χ3 and β12 borophene, much larger than that of H2CO adsorbed on graphene (0.019 e) [38], BN (−0.03 e) [40] and C2N (0.031 e) [19].
To further investigate the formation of the new bonds, the partial density of states (PDOS) of C2H4 and H2CO adsorbed on borophenes are shown in Fig. 3. With the symmetry of the borophenes and C2H4 molecule, one C-B bond for C2H4 adsorbed on χ3 and β12 borophene are considered and the PDOS are shown in Figs. 3a and b, respectively. In Fig. 3a, the major peaks at about −6.5, −4.5, −2.8 and −2.0 eV are contributed by the 2p orbital of C atom of C2H4, while the peaks at about −6.5, −4.5, −2.8 and −2.0 eV are contributed by the 2p orbital of B atom of χ3 borophene, indicating the strong overlap of the 2p orbitals of C and B with the formation of the C-B bond. In addition, considering the geometry analysis that the C atom of adsorbed C2H4 is sp3 hybrid, the C-B bond for C2H4 adsorbed on χ3 borophene is stable σ bond. The same conclusion can be made from the analysis of PDOS of C2H4 adsorbed on β12 borophene as shown in Fig. 3b. Analysis of Figs. 3c and d can find that the steady C-B bonds of H2CO adsorbed borophenes result from the 2p orbitals of C and B atoms resonate at about −7.0, −4.5 and −2.8 eV forχ3 borophene and at about −7.0, −5.0 and −3.1 eV forβ12 borophene. Figs. 3e and f show that the O and B atoms maintain overlap at about −7.0 eV and −2.5 eV forχ3 borophene and at about −7.0, −4.0 and −3.2 eV forβ12 borophene. In other words, H2CO is chemically adsorbed on χ3 and β12 borophene with the formation of two stable chemical bonds of C-B and O-B.

|
Download:
|
| Fig. 3. PDOS of CB bond formed by C2H4 adsorption on χ3 borophene (a) and β12 borophene (b), the PDOS of CB bond formed by H2CO adsorption on χ3 borophene (c) and β12 borophene (d), the PDOS of OB bond formed by H2CO adsorption on χ3 borophene (e) and β12 borophene (f). | |
{kind=link}
It is known that not only the surface structure of adsorbents can be influenced by adsorbed molecules, but also the conductivity, which can be demonstrated by density of states (DOS) in DFT calculation. The DOS of the typical VOCs adsorbed on borophenes are shown in Fig. S4 (Supporting information). In Figs. S4a and b, the DOS of physical-adsorbed VOCs and pure borophenes almost overlap near Fermi level, indicating the ignorable conductivity change of borophenes with the adsorption of VOCs molecules. On the contrary, Figs. S4c and d show the DOS of chemical-adsorbed VOCs and pure borophenes. It can be found that both of χ3 and β12 borophene has a distinct peak near Fermi level, while the value of the peak reduces after adsorbing C2H4 and H2CO, indicating the loss of active electron near Fermi. In other words, the conductivity of borophenes reduces obviously after adsorbing C2H4 and H2CO molecules. These phenomena are corresponding to the Mulliken analysis results which reveal that the adsorbed C2H4 and H2CO can attract electrons from borophenes.
The typical VOCs adsorbing on both χ3 and β12 borophene are investigated by using first-principles calculations. The analysis of adsorption configuration reveals that C2H4 and H2CO can be chemically adsorbed on the two borophenes stably with the chemical bonds formation, while the other VOCs molecules are just adsorbed on borophenes with weak van der Waals force. Besides, the analysis of electron properties for C2H4 and H2CO shows that obvious electron accumulation near the new bonds, obvious electrons transfer from borophenes to the adsorbed VOCs molecules, and apparent orbital overlap occurs between the bonding atoms, indicating that the bonds between the two VOCs molecules and borophenes are steady chemical bond. In addition, the analysis of DOS reveals that the adsorption of C2H4 and H2CO can change the conductivity of bororphene, which is an important character for sensors. Therefore, both χ3 and β12 borophene are able to selective chemical adsorption for C2H4 and H2CO from typical VOCs.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21777033 and 41807191), Science and Technology Planning Project of Guangdong Province (No. 2017B020216003), Natural Science Foundation of Guangdong Province, China (No. 2018A030310524), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01Z032), and the Innovation Team Project of Guangdong Provincial Department of Education (No. 2017KCXTD012).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.026.
| [1] |
T. Lomonaco, E. Manco, A. Corti, et al., J. Hazard. Mater. 394 (2020) 122596. DOI:10.1016/j.jhazmat.2020.122596 |
| [2] |
C. He, J. Cheng, X. Zhang, et al., Chem. Rev. 119 (2019) 4471-4568. DOI:10.1021/acs.chemrev.8b00408 |
| [3] |
S. Scire, L.F. Liotta, Appl. Catal. B 125 (2012) 222-246. DOI:10.1016/j.apcatb.2012.05.047 |
| [4] |
F. Liu, S.P. Rong, P.Y. Zhang, L.L. Gao, Appl. Catal. B 235 (2018) 158-167. DOI:10.1016/j.apcatb.2018.04.078 |
| [5] |
C. Liu, X. Huang, J. Li, Sci. Total Environ. 720 (2020) 137640. DOI:10.1016/j.scitotenv.2020.137640 |
| [6] |
X.X. Feng, H.X. Liu, C. He, et al., Catal. Sci. Technol. 8 (2018) 936-954. DOI:10.1039/C7CY01934C |
| [7] |
D.R. Gentner, S.H. Jathar, T.D. Gordon, et al., Environ. Sci. Technol. 51 (2017) 1074-1093. DOI:10.1021/acs.est.6b04509 |
| [8] |
L. Zhu, D. Shen, K.H. Luo, J. Hazard. Mater. 389 (2020) 122102. DOI:10.1016/j.jhazmat.2020.122102 |
| [9] |
A. Lamplugh, A. Nguyen, L.D. Montoya, Build. Environ. 176 (2020) 106784. DOI:10.1016/j.buildenv.2020.106784 |
| [10] |
G.L. Liu, J.H. Zhou, W.N. Zhao, Z.M. Ao, T.C. An, Chin. Chem. Lett. 31 (2020) 1966-1969. DOI:10.1016/j.cclet.2019.12.023 |
| [11] |
S.H. Zhang, J.P. You, C. Kennes, et al., Chem. Eng. J. 334 (2018) 2625-2637. DOI:10.1016/j.cej.2017.11.014 |
| [12] |
Z.P. Rao, X.F. Xie, X. Wang, et al., J. Phys. Chem. C 123 (2019) 12321-12334. DOI:10.1021/acs.jpcc.9b02093 |
| [13] |
H. Deng, S. Kang, J. Ma, et al., Environ. Sci. Technol. 53 (2019) 10871-10879. DOI:10.1021/acs.est.9b01822 |
| [14] |
X. Zhang, B. Gao, A.E. Creamer, C. Cao, Y. Li, J. Hazard. Mater. 338 (2017) 102-123. DOI:10.1016/j.jhazmat.2017.05.013 |
| [15] |
X.Q. Li, L. Zhang, Z.Q. Yang, et al., Sep. Purif. Technol. 235 (2020) 116213. DOI:10.1016/j.seppur.2019.116213 |
| [16] |
X. Li, H. Zhang, J.F. Wei, et al., Microporous Mesoporous Mater. 303 (2020) 110190. DOI:10.1016/j.micromeso.2020.110190 |
| [17] |
H.D. Ji, P.H. Du, D.Y. Zhao, et al., Appl. Catal. B 263 (2020) 118357. DOI:10.1016/j.apcatb.2019.118357 |
| [18] |
O. Leenaerts, B. Partoens, F.M. Peeters, Phys. Rev. B 77 (2008) 125416. DOI:10.1103/PhysRevB.77.125416 |
| [19] |
Y. Su, Z. Ao, Y. Ji, G. Li, T. An, Appl. Surf. Sci. 450 (2018) 484-491. DOI:10.1016/j.apsusc.2018.04.157 |
| [20] |
B. Liu, W.N. Zhao, Q.G. Jiang, Z.M. Ao, T.C. An, Sustainable Mater. Technol. 21 (2019) e00103. DOI:10.1016/j.susmat.2019.e00103 |
| [21] |
H. Tang, S. Ismail-Beigi, Phys. Rev. B 82 (2010) 115412. DOI:10.1103/PhysRevB.82.115412 |
| [22] |
H. Tang, S. Ismail-Beigi, Phys. Rev. Lett. 99 (2007) 115501. DOI:10.1103/PhysRevLett.99.115501 |
| [23] |
L. Zhang, P. Liang, H.-b. Shu, et al., J. Phys. Chem. C 121 (2017) 15549-15555. DOI:10.1021/acs.jpcc.7b03741 |
| [24] |
T. Tsafack, B.I. Yakobson, Phys. Rev. B 93 (2016) 165434. DOI:10.1103/PhysRevB.93.165434 |
| [25] |
G. Li, Y.C. Zhao, S.M. Zeng, M. Zulfiqar, J. Ni, J. Phys. Chem. C 122 (2018) 16916-16924. DOI:10.1021/acs.jpcc.8b03108 |
| [26] |
H.R. Jiang, W. Shyy, M. Liu, Y.X. Ren, T.S. Zhao, J. Mater. Chem. A 6 (2018) 2107-2114. DOI:10.1039/C7TA09244J |
| [27] |
B. Feng, J. Zhang, Q. Zhong, et al., Nat. Chem. 8 (2016) 563-568. DOI:10.1038/nchem.2491 |
| [28] |
X. Wu, J. Dai, Y. Zhao, et al., ACS Nano 6 (2012) 7443-7453. DOI:10.1021/nn302696v |
| [29] |
T. Liu, Y. Chen, M. Zhang, et al., AIP Adv. 7 (2017) 125007. DOI:10.1063/1.5005959 |
| [30] |
C. Dang, F. Sun, H. Jiang, et al., J. Hazard. Mater. 400 (2020) 123225. DOI:10.1016/j.jhazmat.2020.123225 |
| [31] |
H. Ji, T. Wang, T. Huang, B. Lai, W. Liu, J. Cleaner Prod. 278 (2021) 123924. DOI:10.1016/j.jclepro.2020.123924 |
| [32] |
L. Jeloaica, V. Sidis, Chem. Phys. Lett. 300 (1999) 157-162. DOI:10.1016/S0009-2614(98)01337-2 |
| [33] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [34] |
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799. DOI:10.1002/jcc.20495 |
| [35] |
B. Delley, Phys. Rev. B 66 (2002) 155125. DOI:10.1103/PhysRevB.66.155125 |
| [36] |
C. Özdoğan, S. Mukhopadhyay, W. Hayami, et al., J. Phys. Chem. C 114 (2010) 4362-4375. DOI:10.1021/jp911641u |
| [37] |
F. Perrot, Phys. Rev. B 23 (1981) 2004-2010. DOI:10.1103/PhysRevB.23.2004 |
| [38] |
M. Chi, Y.P. Zhao, Comput. Mater. Sci. 46 (2009) 1085-1090. DOI:10.1016/j.commatsci.2009.05.017 |
| [39] |
X.X. Deng, X.Y. Liang, S.P. Ng, C.M.L. Wu, Appl. Surf. Sci. 484 (2019) 1244-1252. DOI:10.1016/j.apsusc.2019.04.175 |
| [40] |
S.Y. Liu, X.Q. Jiao, G.Y. Zhang, Chem. Phys. Lett. 726 (2019) 77-82. DOI:10.1016/j.cplett.2019.04.041 |