 2021, Vol. 32
2021, Vol. 32 


The organocatalyzed reactions for the asymmetric synthesis of C-C and C-X bonds have gained extensive research interest in organic chemistry, which provide a complementary strategy to the metal- and enzyme-catalyzed reactions [1, 2]. Over past decades, a wide range of the organocatalysis, such as Brønsted acids, phosphines, N-heterocyclic carbenes, prolines and thiourea catalysts, has been developed [2]. In this respect, considerable research efforts have been devoted to the phosphine catalysts, which have shown great potential for the cycloaddition and addition reactions [3]. For instance, Trost and co-workers in 1994 developed the phosphine-catalyzed "umpolung" addition [4]. Lu and co-workers in 1995 reported an early discovery on the phosphine-catalyzed [3+2] annulation between 2, 3-butadienoates or 2-butynoates with electron-deficient olefins [5], which has become one of the most powerful tools for the construction of the five-membered-ring carbo- and heterocycles.
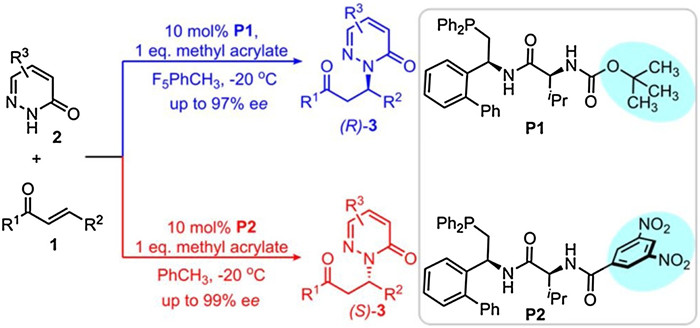
Despite the significance progress has been witnessed, the development of the enantiodivergent phosphine-catalyzed reactions remains a central challenge in this field [6]. In this context, Zhang and co-workers very recently reported an elegant example of the dipeptide phosphine-catalyzed hydroamination of enones 2 with pyridazinones 1 (Scheme 1) in high yields and with good enantioselectivity [7]. The salient feature of the reaction is that both enantiomers of the addition products 3 can be obtained by the careful selection of the dipeptide phosphine catalyst. It was found that with the dipeptide phosphine P1, the addition of pyridazinones 1 to enones 2 can give the products (R)- 3 with the excellent enantioselectivity up to 97% ee. However, by using the catalyst P2, wherein the substituent of the amide moiety of P1 was changed from the OBoc group to the 3, 5-dinitrobenzyl group, the reversed enantioselectivity was observed and the products (S)- 3 were obtained with the enantioselectivity up to 99% ee. The reaction represents thus a rare case of the enantiodivergent phosphine-catalyzed reactions without changing any stereocenter of the catalyst.

|
Download:
|
| Scheme 1. Dipeptide phosphine-catalyzed enantiodivergent hydroamination reaction. | |
To gain insight into the detailed reaction mechanism and the origins of this unprecedented catalyst-controlled enantioselectivity, density functional theory (DFT) calculations at the M06-2X/6-311+G (d, p)-SMD//M06-2X/6-31G (d)-SMD level of theory were performed (see Supporting information for computational details) [8, 9]. The experimentally used β-trifluoromethylated enone 1a and pyridazinone 2a were selected as the model substrates. Based on our computational study, the detailed reaction mechanism of the phosphine-catalyzed hydroamination is shown in Scheme 2. The reaction begins with the nucleophilic attack of the phosphine catalyst to methyl acrylate, delivering a zwitterionic intermediate INT1, which then undergoes the protonation with pyridazinone 2 to give ionic pair intermediate INT2. The subsequent Michael addition between INT2 and enone 1 leads to intermediate INT3. Finally, the catalytic cycle is closed by the protonation of intermediate INT3 with pyridazinone 2 to release the hydroamination product 3 and regenerate INT2. The computations show that in both P1- and P2-catalyzed reactions, the Michael addition constitutes the enantioselectivity-determining step of the overall catalytic cycle.

|
Download:
|
| Scheme 2. Proposed reaction mechanism based on DFT calculations. | |
The calculated energy profile of the P1-catalyzed reaction is given in Fig. 1a (see Supporting information for the optimized geometries of the selected transition states). The computations show that the nucleophilic attack of the P atom of the catalyst to the methyl acrylate takes place via transition state P1-TS1 with an energy barrier of 16.6 kcal/mol, generating zwitterionic intermediate P1-INT1. Then, this intermediate undergoes the protonation with pyridazinone 2a via transition state P1-TS2 leading to ionic pair intermediate P1-INT2. Upon formation of P1-INT2, two possible modes of the Michael addition, namely attack of the Re and Si faces of enone 1a, were both considered, which were found to occur via transition states P1-TS3-Re and P1-TS3-Si, respectively. Finally, the protonation of the resulting intermediates P1-INT3-Re and P1-INT3-Si with pyridazinone 2a to regenerate P1-INT2 and release the hydroamination products (R)-3a and (S)- 3a, respectively. The results show that the Re face attack through transition state P1-TS3-Re is lower in energy than the Si face attack via transition state P1-TS3-Si by 2.7 kcal/mol, which is in good agreement with the experimentally observed 95% ee in favor of (R)- 3a.

|
Download:
|
| Fig. 1. Calculated energy profiles of the P1- and P2-catalyzed hydroamination of 1a with 2a (R = 4 - ClC6H4). | |
The P2-catalyzed reaction follows the same reaction mechanism as established for the case of the P1 catalyst (Fig. 1b and see Supporting information for the optimized geometries of the selected transition states). However, the catalyst was indeed found to have a significant impact on switching the enantioselectivity. With the P2 catalyst, the Re face attack via transition state P2-TS3-Re is disfavored over the Si face attack via transition state P2-TS3-Si by 2.9 kcal/mol, being in accordance with the experiments that the reversed enantioselectivity was observed for the P2-catalyzed reaction. Therefore, the calculations reproduced quite well the experimentally observed catalyst-controlled enantioselectivity.
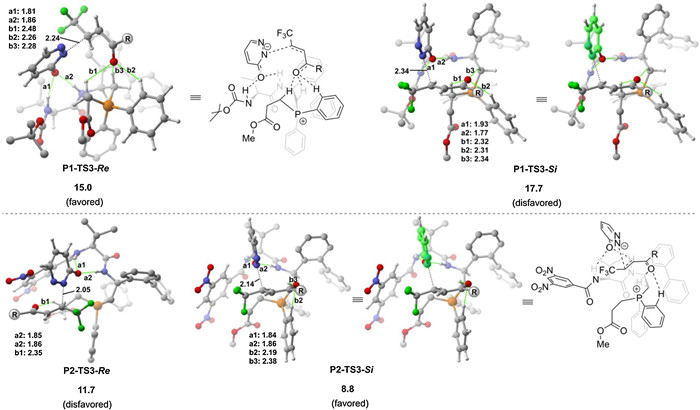
The optimized geometries of the enantioselectivity-determining transition states are given in Fig. 2. The computations show that a number of the N-H···O hydrogen-bonding interactions with the pyridazinone moiety and the C-H···O hydrogen-bonding interactions with the β-trifluoromethylated enone moiety are present in enantioselectivity-determining Michael addition transition states. For the P1-catalyzed reaction, the distortion/interaction analysis [10] of P1-TS3-Si and P1-TS3-Re was conducted to gain insights into the origins of the selectivity. As depicted in Table 1, the results show that the distortion energies of 1a (ΔE1a) for both transition states are nearly identical (4.9 and 4.1 kcal/mol). The selectivity is mainly determined by the difference in the distortion energy of P1-INT2. ΔEP1-INT2 of P1-TS3-Si was calculated to be higher than that of P1-TS3-Re by 3.5 kcal/mol (8.2 kcal/mol versus 4.7 kcal/mol), which consequently results in the energy of P1-TS3-Si being higher than P1-TS3-Re. The origins of this is likely due to that for the Si face attack, the pyridazinone moiety of P1-INT2 has to be distorted to achieve the geometric structure of the P1-TS3-Si. On the other hand, the relative orientation of the pyridazinone moiety can readily undergo the Re face attack.

|
Download:
|
| Fig. 2. Optimized geometric structures of the Michael addition transition states (R=4-ClC6H4). Energies and bond distances are given in kcal/mol and Å, respectively. For the sake of clarity, some irrelevant hydrogen atoms were omitted. | |
|
|
Table 1 Distortion/interaction analysis (kcal/mol) of P1-TS3-Si and P1-TS3-Re.a |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
By changing the catalyst to P2, the geometric structure of the Re face attack transition state P2-TS3-Re was found to be quite different to that of P1. We have also considered the Re face attack transition state with P2 with the geometric structure similar to that of P1-TS3-Re. However, due to the steric repulsion between the 3, 5-dinitrobenzyl group and pyridazinone, the energy of the corresponding transition state was calculated to be 2.8 kcal/mol higher in energy than P2-TS3-Re (see Supporting information for details). The comparison of the optimized geometries of P1-TS3-Si and P2-TS3-Si shows that the substituent of the amide moiety has a significant impact on the relative strength of the N-H···O hydrogen-bonding interactions. In P1-TS3-Si, the N-H···O hydrogen-bonding interaction a1 is weaker than a2 (1.93Å versus 1.77Å and ENBO = 8.41 kcal/mol of a1 versus ENBO = 15.52 kcal/mol of a2). While in P2-TS3-Si, due to the strong electron-withdrawing character of the 3, 5-dinitrobenzyl group, the N-H···O hydrogen-bonding interaction a1 was found to be slightly stronger than a2 (1.84Å versus 1.86Å and ENBO = 13.10 kcal/mol of a1 versus ENBO = 10.21 kcal/mol of a2). Very importantly, the difference in the relative strength of the N-H···O hydrogen-bonding interactions makes the orientation of the pyridazinone being quite different in two transition states. In P1-TS3-Si, the pyridazinone was found to orient away from the β-trifluoromethylated enone moiety, where in P2-TS3-Si, the pyridazinone was found to orient toward to the β-trifluoromethylated enone moiety. As a result, the distance of the forming C-N bond in P2-TS3-Si was found to be much shorter than in P1-TS3-Si (2.14Å versus 2.34Å), which may facilitate the Si face attack and thus enable the enantioselectivity switch upon change of chiral dipeptide phosphine catalyst. This argument is also in accordance with the control experiments that the second N2-H of chiral dipeptide phosphine catalyst is crucial to reverse the enantioselectivity [7].
To summarize, we have herein presented a mechanistic study on the dipeptide phosphine-catalyzed hydroamination of enones with pyridazinones by means of DFT calculations. The computations show that the enantioselectivity of the reaction is determined by the Michael addition between enone and the ionic pair intermediate, generated by the initial nucleophilic attack/protonation. The experimentally-observed catalyst-controlled enantiodivergence was reproduced quite well by the calculations. It was found that a number of the N-H···O hydrogen-bonding interactions with the pyridazinone moiety and the C-H···O hydrogen-bonding interactions with the β-trifluoromethylated enone moiety are present in the Michael addition transition states. The electronic character of the substituent of the amide moiety of the dipeptide phosphine has a significant impact on the relative strength of the N-H···O hydrogen-bonding interactions, which was found to affect the Si face attack transition state, leading to the enantioselectivity switch upon change of chiral dipeptide phosphine catalyst.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 22073066, 21503143 and 21975179) and the Natural Science Foundation of Tianjin (No. 16JCQNJC05600).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.049.
| [1] |
(a) S. Mukherjee, J.W. Yang, S. Hoffmann, B. List, Chem. Rev. 107 (2007) 5471-5569; (b) Y. Qin, L. Zhu, S. Luo, Chem. Rev. 117 (2017) 9433-9520; (c) L. Dai, S. Ye, Chin. Chem. Lett. 32 (2021) 660-667; (d) E.Z. Lin, Y. Xu, K. Ji, L.W. Ye, Chin. Chem. Lett. 32 (2021) 954-962; (e) W. Cao, X. Liu, X. Feng, Chin. Chem. Lett. 29 (2018) 1201-1208. |
| [2] |
(a) Y.C. Fan, O. Kwon, Chem. Commun. 49 (2013) 11588-11619; (b) B.J. Cowen, S.J. Miller, Chem. Soc. Rev. 38 (2009) 3102-3116; (c) H. Guo, Y.C. Fan, Z. Sun, Y. Wu, O. Kwon, Chem. Rev. 118 (2018) 10049-10293; (d) H. Ni, W.L. Chan, Y. Lu, Chem. Rev. 118 (2018) 9344-9411. |
| [3] |
(a) X. Han, W.L. Chan, W. Yao, Y. Wang, Y. Lu, Angew. Chem. Int. Ed. 55 (2016) 6492-6496; (b) Y. Gu, P. Hu, C. Ni, X. Tong, J. Am. Chem. Soc. 137 (2015) 6400-6406; (c) B. Huang, C. Li, H. Wang, et al., Org. Lett. 19 (2017) 5102-5105; (d) W. Zhou, H. Wang, M. Tao, et al., Chem. Sci. 8 (2017) 4660-4665; (e) W. Yao, Z. Yu, S. Wen, et al., Chem. Sci. 8 (2017) 5196-5200; (f) H. Ni, Z. Yu, W. Yao, et al., Chem. Sci. 8 (2017) 5699-5704; (g) J. Zhang, H.H. Wu, J. Zhang, Org. Lett. 19 (2017) 6080-6083; (h) L. Zhang, H. Liu, G. Qiao, et al., J. Am. Chem. Soc. 137 (2015) 4316-4319; (i) H. Wang, L. Zhang, Y. Tu, et al., Angew. Chem. Int. Ed. 57 (2018) 15787-15791. |
| [4] |
B.M. Trost, C.J. Li, J. Am. Chem. Soc. 116 (1994) 3167-3168. DOI:10.1021/ja00086a074 |
| [5] |
C. Zhang, X. Lu, J. Org. Chem. 60 (1995) 2906-2908. DOI:10.1021/jo00114a048 |
| [6] |
(a) C.E. Henry, Q. Xu, Y.C. Fan, et al., J. Am. Chem. Soc. 136 (2014) 11890-11893; (b) Z. Wang, T. Wang, W. Yao, Y. Lu, Org. Lett. 19 (2017) 4126-4129; (c) T. Wang, Z. Yu, D.L. Hoon, et al., Chem. Sci. 6 (2015) 4912-4922; (d) A.J. Smaligo, S. Vardhineedi, O. Kwon, ACS Catal. 8 (2018) 5188-5192; (e) E. Li, H. Jin, P. Jia, X. Dong, Y. Huang, Angew. Chem. Int. Ed. 55 (2016) 11591-11594. |
| [7] |
H. Wang, X. Li, Y. Tu, J. Zhang, iScience (2020) 101138. DOI:10.1016/j.isci.2020.101138 |
| [8] |
(a) H. Li, X. Hong, Chin. Chem. Lett. 29 (2018) 1585-1590; (b) P.H.Y. Cheong, C.Y. Legault, J.M. Um, N. Çelebi-Ölçüm, K.N. Houk, Chem. Rev. 111 (2011) 5042-5137; (c) C.X. Cui, C. Shan, Y.P. Zhang, et al., Chem. Asian J. 13 (2018) 1076-1088. |
| [9] |
(a) Y. Xia, Y. Liang, Y. Chen, et al., J. Am. Chem. Soc. 129 (2007) 3470-3471; (b) H. Chen, L. Zhu, K. Zhong, et al., Chin. Chem. Lett. 29 (2018) 1237-1241; (c) Y. Wang, Y. Lan, Chin. Chem. Lett. 31 (2020) 736-738; (d) Y. Wang, M. Tang, Y. Wang, D. Wei, J. Org. Chem. 81 (2016) 5370-5380; (e) T. Liu, S. Han, Y. Li, S. Bi, J. Org. Chem. 81 (2016) 9775-9784; (f) Z. Yu, Z. Jin, M. Duan, et al., J. Org. Chem. 83 (2018) 9729-9740; (g) B. Bhaskararao, G. Jindal, R.B. Sunoj, J. Org. Chem. 82 (2017) 13449-13458; (h) G. Huang, C. Diner, K.J. Szabó, F. Himo, Org. Lett. 19 (2017) 5904-5907. |
| [10] |
(a) F.M. Bickelhaupt, K.N. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070-10086; (b) D.H. Ess, K.N. Houk, J. Am. Chem. Soc. 129 (2007) 10646-10647; (c) X. Hong, Y. Liang, K.N. Houk, J. Am. Chem. Soc. 136 (2014) 2017-2025; (e) S. Liu, Y. Lei, X. Qi, Y. Lan, J. Phys. Chem. A 118 (2014) 2638-2645; (f) G. Huang, M. Kalek, R.Z. Liao, F. Himo, Chem. Sci. 6 (2015) 1735-1746; (g) X. Zhang, H. Zou, G. Huang, ChemCatChem 8 (2016) 2549-2556; (h) J. Yu, S. Zhang, X. Hong, J. Am. Chem. Soc. 139 (2017) 7224-7243; (i) H. Wu, X. Li, X. Tang, C. Feng, G. Huang, J. Org. Chem. 83 (2018) 9220-9923; (j) H. Zou, Z.L. Wang, Y. Cao, G. Huang, Chin. Chem. Lett. 29 (2018) 1355-1358. |