 2021, Vol. 32
2021, Vol. 32 


Cascade cycloisomerization reactions of unactivated 1, n-enynes or diynes are powerful synthetic strategies for fused polyheterocycles in efficient and green synthetic protocols [1]. However, these types of reactions typically necessitate high temperatures (> 180 ℃) [2] or visible light [3] to induce the initial radical formation. Transition-metal catalysts can be employed for multiple steps in a cascade cyclization protocol for products with significant molecular complexity. A wide range of important cyclic derivatives, such as polysubstituted benzenes [4], naphthyl ketones [5], fulvenes [6], benzofluorenes [7], and indenes [8] were chemo- and stereo- selectively produced from 1, n-enynes or diynes with various transition-metal catalyzed conditions.
The catalytic halocyclization reactions of 1, n-enynes or diynes are useful tools for the preparations of halo-substituted polyheterocycles. For example, in 2009, Song and Dong reported a Pd-catalyzed intramolecular [3+2] carboesterification reaction of propiolic acids and unactivated olefins to generate a tricyclic fused ring system (Scheme 1a) [9]. Our group recently developed the transition-metal-catalyzed electrophilic cycloisomerization reactions of benzo-fused 1, 6-diynols or triynols with N-halosuccinimides (NXS, X=I, Br) [10]. Under the catalysis of a transition-metal catalysts, functionalized carbo(hetero)cyclic frameworks such as benzo[a]fluorenes, benzo[b]fluorenones, or naphthalenes were efficiently and chemoselectively produced under gentle reaction conditions. While the success of these examples referred to the halocyclizations of the ortho-fused aryl 1, n-enynes or diynes, instances for the halocyclization reactions of the unactivated acyclic 1, n-diynes are much more challenging. This is because the remote π-bonds have a very wide free rotational angle in these "unlocked" acyclic linear systems; therefore, the reactivity and the chemo- and/or regioselectivity are much more difficult to control.

|
Download:
|
| Scheme 1. Halocyclization of unactivated 1, 6-diynes and the further derivation to 1, 3-dienes. | |
To achieve a satisfactory endeavor in the non-conjugated acyclic unactivated diynes, Snyder and co-workers in 2015 reported that a stoichiometric amount of GaI3-promoted cyclization of 1, 6-diynes to obtain aza-heterocyclic vinyl iodides, that could undergo an in situ Friedel-Crafts cyclization to give the iodoindenopyridine derivatives (Scheme 1b) [11]. We herein disclose a Pd/Cu co-catalyzed halocyclization reaction of acyclic 1, 6-diynes with NBS, which produces stereo-defined dibrominated (E)-3, 6-dihydro-2H-pyrans in good yields (Scheme 1c). The resulting dibromo products could undergo ring-opening reactions with B(C6F5)3/HSiEt3 to stereo selectively give substituted 1, 3-dienes, which are highly conjugated polyene structural motifs and useful building blocks in the synthesis of natural products such as β-carotene, vitamin A, and other naturally occurring retinoids (Scheme 1d) [12]. The dibromo-substituted tetrahydropyridines and 3-methylene cyclohexenes can also be stereoselectively prepared in good yields from this reaction.
We began to survey the reaction by utilizing 1, 6-diynyl ether 1a and 2.1 equiv. of N-bromosuccinimide (NBS) as the substrates (Table 1). After an extensive examination of the reaction parameters, we found optimized reaction conditions consisting of PdBr2 (10 mol%), and 10 mol% of CuBr2 as an effective additive in 0.1 mol/L of THF at 130 ℃. The desired product 2a was isolated in 83% yield under this "standard condition" (entry 1). The Pd-catalyst was necessary, because no product was detected in the absence of any Pd-catalyst (entries 2 and 3). The Pd salt with halide anion (PdCl2, PdBr2 or Pd(PPh3)2Cl2 alone) was effective for the reaction. Without CuBr2, however, an inert reaction was observed when Pd(OAc)2 and Pd(dba)2 were utilized (entries 4–7). These results indicated that the halide anion was important for the initiation of the reaction. Interestingly, CuBr2 worked as a Lewis acid could dramatically promote the reaction (entry 4 vs. 1).
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
Informed by this result, we next screened of other Lewis acids as promoters, including FeBr2, FeBr3, CoBr2, NiBr2 and ZnBr2. However, none of the other Lewis acids offered any satisfactory results (entries 8–12). A smooth conversion was observed when CuBr was utilized, and the desired product 2a could be isolated in 72% yield (entry 13). In this solvent, the reaction worked well in 1, 2-dichloroethane to give the desired product 2a in 77% yield (entry 14). However, switching to other laboratory solvents such as toluene, 1, 4-dioxane, or DMF did not increase the efficiency (entries 15–17). Poor results were observed when the loading of PdBr2 or CuBr2 was lowered to 5.0 mol% (entries 18 and 19). In addition, the yield of product 2a decreased from 130 ℃ to 105 ℃ with a reaction time of 12h (entry 20).
Once the reaction conditions were established, the scope of this reaction was next studied. Scheme 2 shows a variety of bromide functionalized (E)-3, 6-dihydro-2H-pyrans with exocyclic double bond appendages. These were readily allowed when NBS was utilized as the halogen source. The reaction was quite general with respect to variation of the aryl groups on the alkyne moieties that attached with different electronic natures. Diverse series of products 2b-2n were regio-specifically generated in good yields. The electron-neutral phenyl substituted 1, 6-diynyl ether 2b was produced in 69% yield. When the R group was attached at the phenyl moiety of 1, 6-diynyl ether 1, electron-donating alkyl substituents such as Me and tBu proceeded smoothly. Note that nearly comparable yields of products 2c, 2d, and 2e were also obtained, suggesting that the location of the methyl group at the para-, meta- or ortho-positions of phenyl ring had little impact on the efficiency. The corresponding product 2f was obtained in 65% yield when R was a bulky tertiary butyl group.

|
Download:
|
| Scheme 2. Reaction scope studies. Reaction conditions: 1, 6-diynyl ether 1 (0.40 mmol), NBS (0.82 mmol), PdBr2 (10 mol%), CuBr2 (10 mol%), THF (4.0mL), 130 ℃, 8h. Toluene (0.1 mol/L), 80 ℃ instead of THF (0.1 mol/L), and 130 ℃. | |
{kind=link}
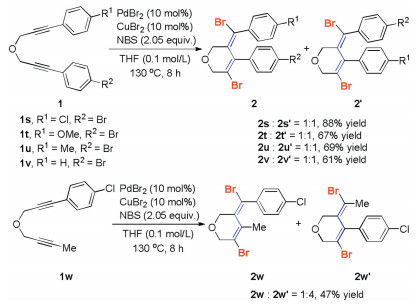
Products 2e and 2i were obtained as a mixture of atropisomers as indicated by their proton NMR, they had axial chiral properties at room temperature (Scheme 3). When the aryl substituent attached to the alkyne terminus became more electron deficient, the reaction efficiency increased dramatically versus with the aryl substituent attached to the electron-rich alkyne terminus (Scheme 2). The presence of a mild electron-withdrawing fluorine atom at para- or meta-positions of the phenyl moiety were well tolerated in the reaction, affording the corresponding products 2g and 2h in 76% and 85% yields, respectively. Encouraged by these results, we expanded the reaction to the other halogen substituted electron-deficient arenes. The use of chlorine-containing aryl substituents of 1, 6-diynyl ethers also resulted in good yields of the desired products (2j and 2k). The substrate that contained a bromide atom (R=Br) at the 4-Ph moiety underwent the reaction to give product 2l in good yield. A nice reaction for the 2-phenyl substituted substrate produced the expected product 2m in 84% yield. The structure of product 2m was established unambiguously by single-crystal X-ray crystallography diffraction analysis (CCDC: 1985748). The synthetically useful cynano group could be tolerated under slightly modified reaction conditions, providing the desired products 2o and 2p in modest yields. The ester group was tolerated, and 2q was isolated in 86% yield in toluene as the solvent at 80 ℃. Aryl, alkyl, and heterocyclic thienyl groups could be placed at the triple terminal position to provide the corresponding product 2r. Unfortunately, strong electron-withdrawing groups such NO2 and CF3, are not compatible with the reaction.

|
Download:
|
| Scheme 3. Origination of the atropisomer 2e. | |
{kind=link}
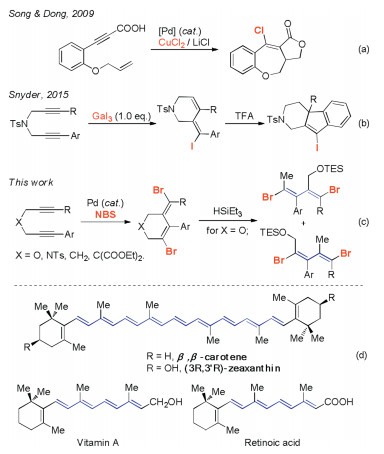
The cyclization regioselectivity of 1, 6-diynyl ether 1 bearing two different functional groups was next studied under standard reaction conditions (Scheme 4). For the 1, 6-diyne 1t with an electron-donating OMe group and an electron-withdrawing Br group substituted phenyl substituents at each alkyne terminus, two isomeric products 2t and 2t' were isolated as an inseparable mixture in 67% yield. The observed molecular ratio of 2t/2t' was 1:1 as indicated by 1H NMR, showed that the electronic nature of the phenyl ring had less impact on the site selectivity on the alkyne carbon atoms. The preference for cyclization of 1, 6-diynyl ether 1u, which individually bears a methyl group and a bromide group at the phenyl moieties, is also not obvious. The two resulting isomers 2u and 2u' = 1:1 were obtained in a 1:1 molecular ratio. Similar results were observed for the other 1, 6-diynyl ethers 1s and 1v, their corresponding products were isolated in 61%–88% yields. An example of this exception is the cyclization reaction of w, which contains a Me group and a 4-Cl phenyl group at each of the alkyne terminus. The two cyclic isomers 2w and 2w' were produced in a molecular ratio of 1:4 in 47% yield.

|
Download:
|
| Scheme 4. Substrate scope studies. | |
{kind=link}
It should be noted we have tried the reaction of 1, 6-diynyl ethers 1a with N-chlorosuccinimide (NCS) or N-iodosuccinimide (NIS) that instead of NBS to participate in the reaction. In the case of NCS, the conversion was rather complex but there was no desired product detected. Unfortunately, in the reaction 1a with of NIS, no reaction was observed under the standard conditions.
To further explore the scope of this halocyclization reaction, diversified unactivated 1, 6-diynes 3, bearing a different –X moiety were evaluated (Scheme 5). Overall, a series of dibrominated aza- or carbocycles were successfully introduced affording the anticipated 3-methylenetetrahydropyridines or 3-methylenecyclohexenes 4a-4h in 39%–80% yields. Similar to the standard 1, 6-diynyl ether 1a, compound 3a that was prepared from bis(3-phenylprop-2-yn-1-yl)amine proceeded in the Pd/Cu co-catalyzed reaction smoothly for the generation of product 4a in 69% yield. Treatment of substrates 3b or 3c bearing a Me or a Cl group at the phenyl moeity afforded the corresponding products 4b and 4c in good yields, respectively. The carbon chain 1.6-diyne 3d-3f, which derived from diethyl malonate, was successfully transformed into their corresponding products 4d-4f in 50%–69% yields. Gratifyingly, the acyclic carbon chain 1.6-diyne 4g and 4h could also be compatible to the halocyclization protocol, providing products 4g and 4h in serviceable yields, respectively. Interestingly, when compound 3-(prop-2-yn-1-yloxy)prop-1-yne, which bearing two hydrogen atoms at the alkyne moieties, was utilized as the starting material, the annulation occurred at the sterically less hindered terminal carbon, thus to generate the azepine product 4i in 31% yield. However, for the dialkyl substituted substrate 3j, which bearing two butyl groups at the 1, 7-terminal positions of 1, 6-diyne, the reaction was rather complex and almost no desired product 4j was observed.

|
Download:
|
| Scheme 5. Substrate scope studies. Toluene (0.1 mol/L), 80 ℃ instead of THF (0.1 mol/L), 130 ℃. | |
{kind=link}
Unlike the previous reports that utilize a 2- to 3-folds stoichiometric excess of metal halides [9, 11, 13] to complete the catalytic cycle, this work only uses a catalytic amount of MBr2 (M=Pd, Cu) in this reaction. Thus, the mechanism of the current reaction should be conceptually different from prior works. To shed light on the reaction, control experiments were conducted. When typical radical scavengers, such as TEMPO, BHT or styrene was added, the reaction became rather sluggish, and product 2a was formed in very poor yields (Scheme 6). These results indicated that a radical-process should be involved in this tandem annulation reaction. To further confirm an organic radical species is involved in the overall process, electron paramagnetic resonance (EPR) experiment was conducted (Fig. 1). When compound 1a was employed as a starting material under the standard conditions for 20min, we successfully observed a radical signal by using DMPO as a trapping agent. This result suggested that the organic radical mediated annulation is involved in this annulation sequences.

|
Download:
|
| Scheme 6. Mechanistic studies. TEMPO=2, 2, 6, 6-tetramethyl-1-piperidinyloxy; BHT=2, 6-di-tert-butyl-4-methylphenol. | |
{kind=link}

|
Download:
|
| Fig. 1. EPR studies by using DMPO as an additive. DMPO=5-dimethyl-1-pyrroline N-oxide. | |
{kind=link}
We proposed a plausible mechanism based on the observations above and our recent studies on the NXS-mediated halocyclization reactions, (Scheme 7) [10]. Here, PdBr2 was the essential catalyst (Table 1, entries 1–3 vs. 5), and we realized that the coordination of PdII and nucleophilic attack of Br− should be the initial step for this cascade transformation. Thus, the coordination reaction of the palladium cationic species to the alkyne functional groups might occur first to form a Pd-π-alkyne complex A [14]. The alkyne group was activated through this coordination to be more electrophilic. It then underwent a trans-bromopalladation reaction to deliver a brominated vinyl palladium species B [15]. The resulting intramolecular syn-carbopalladation to the tethered alkyne moiety can be envisioned to form the cyclic intermediate C [16]. Subsequently, cis/trans isomerization of the emerging Pd-vinyl species through a η2-vinyl transition state D with regio- and stereoselective manner may give formal anti-carbopalladation complex E [17]. The PdII-catalyst would be oxidized by a bromide radical that originated from homolysis of NBS at heated temperature, to produce a hypervalent PdIII-complex F. The subsequent reductive elimination (R.E.) of F delivered product 2 and low-valent PdI-species. In the further oxidations by bromide radical, this low-valent PdI-species can be oxidized to regenerate the active PdII-catalyst to complete the catalytic cycle, along with the generation of stoichiometric amounts of Br− anions. The succinimide radical dimer G could be detected by HRMS ((M+H)+: 197.0552).

|
Download:
|
| Scheme 7. Proposed mechanism. | |
{kind=link}
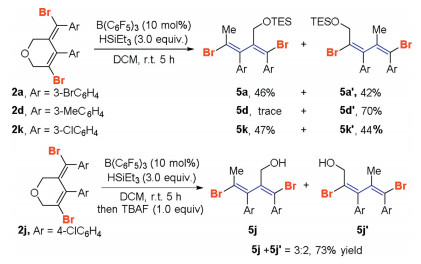
The 1, 3-diene structural motifs are widely found in natural products and pharmaceutical substances, rendering the scaffold very attractive. Thus, to utilize the diene modular structure of the resulting cyclic ether (E)-3, 6-dihydro-2H-pyrans 2, we next studied the B(C6F5)3-catalyzed ring opening reaction of the C-O bonds using HSiEt 3 as a reducing agent to prepare stereo-defined dibromo substituted 1, 3-dienes [18]. Indeed, a series of brominated 1, 3-dienes 5 and 5' were generated in excellent stereoselectivity in overall good yields (Scheme 8). For instance, the B(C6F5)3-catalyzed reaction of 2a with 3.0 equiv. of HSiEt3 in dichloromethane proceeded smoothly to give 1, 3-dienes 5a and 5a' in 46% and 42% yields, respectively. The two isomeric products 5a and 5a' were separable on a silica gel column. Compound 2k participated in the reaction to give products 5k and 5k' in 47% and 44% yields, respectively. Product 2d contains a 3-methylphenyl moiety, and the product 1, 3-diene 5d' was isolated in 70% yield. Moreover, in the B(C6F5)3-catalyzed ring-opening reaction of 2j, an inseparable mixture of 1, 3-diene alcohol 5j and 5j' was produced in 73% yield following by treatment of 1.0 equiv. of TBAF. The dibromide substituted dienes might be served as a valuable building block for the preparation of π-conjugated alkenes.

|
Download:
|
| Scheme 8. Stereoselective preparation of the1, 3-dienes 5. TES=triethylsilyl. | |
{kind=link}
In conclusion, we developed a novel Pd/Cu co-catalyzed cascade halocyclization reaction, consisting of a trans-halopalladation, formal anti-carbopalladation, folllowed by a terminating bromide radical mediate oxidative addition/reductive elimination sequences. A series of stereo-defined dibromo-substituted heterocyclic molecules or methylene cyclohexenes were produced in good to excellent yields. Further B(C6F5)3-catalyzed ring opening reaction of the dihydropyrans afforded a series of synthetically useful brominated 1, 3-dienes with excellent stereoselectivity. Mechanistically, a trans-halopalladation/formal anti-carbopalladation/and a bromide radical promoted PdII-PdIII-PdI-PdII catalytic cycle were proposed to be involved for the formation of these dibromo- substituted products.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Natural Science Foundation of China (No. 21961015) and the Natural Science Foundation of Jiangxi Province (No. 20202ACBL203005) for financial support. Z. Chen was grateful for the Open Project Program of Polymer Engineering Research Center, Jiangxi Science & Technology Normal University (No. KFGJ18014).
| [1] |
(a) I.G. Stara, I. Stary, Acc. Chem. Res. 53 (2020) 144-158; (b) L. Chung, C. Wong, Chem. Eur. 25 (2019) 2889-2897; (c) Y. Hu, M. Bai, Y. Yang, Q. Zhou, Org. Chem. Front. 4 (2017) 2256-2275; (d) C. Shu, L. Li, T. Tan, D. Yuan, L. Ye, Sci. Bull. 62 (2017) 352-357; (e) T. Müller, Metal Catalyzed Cascade Reactions, Vol. 19, Springer, Berlin/Heidelberg, 2006, pp. 149-205. |
| [2] |
(a) D. Rodríguez, M. Martínez-Esperón, A. Navarro-Vázquez, et al., J. Org. Chem. 69 (2004) 3842-3848; (b) R.C. Burell, K.J. Daoust, A.Z. Bradley, K.J. Dirico, P.R. Johnson, J. Am. Chem. Soc. 118 (1996) 4218-4219; (c) D. Rodríguez, A. Navarro, L. Castedo, D. Domínguez, C. Saá, Org. Lett. 2 (2000) 1497-1500; (d) A.G. Myers, E.Y. Kuo, N.S. Finney, J. Am. Chem. Soc. 111 (1989) 8057-8059. |
| [3] |
(a) M. Huang, C. Zhu, C. He, et al., Org. Chem. Front. 5 (2018) 1643-1650; (b) M. Huang, Y. Zhu, W. Hao, et al., Adv. Synth. Catal. 359 (2017) 2229-2234. |
| [4] |
S. Saito, Y. Yamamoto, Chem. Rev. 100 (2000) 2901-2915. DOI:10.1021/cr990281x |
| [5] |
(a) J. Zhao, C.O. Hughes, F.D. Toste, J. Am. Chem. Soc. 128 (2006) 7436-7437; (b) C. Oh, A. Kim, W. Park, D. Park, N. Kim, Synlett 17 (2006) 2781-2784. |
| [6] |
(a) Y. Chen, M. Chen, Y. Liu, Angew. Chem. Int. Ed. 51 (2012) 6493-6497; (b) Q. Xiao, H. Zhu, G. Li, Z. Chen, Adv. Synth. Catal. 356 (2014) 3809-3815; (c) B.M. Trost, T. Michael, T. Rudd, J. Am. Chem. Soc. 127 (2005) 4763-4776. |
| [7] |
(a) J. Zhang, H. Wang, S. Ren, W. Zhang, Y. Liu, Org. Lett. 17 (2005) 2920-2923; (b) W. Hui, Y. Zhou, Y. Dong, et al., Green Energy Environ. 4 (2019) 53-59; (c) J. Zhang, H. Zhang, D. Shi, H. Jin, Y. Liu, Eur. J. Org. Chem. 33 (2016) 5545-5558. |
| [8] |
(a) D. Leboeuf, A. Simonneau, C. Aubert, et al., Angew. Chem. Int. Ed. 50 (2011) 6868-6871; (b) J. Lian, P. Chen, Y. Lin, H. Ting, R.S. Liu, J. Am. Chem. Soc. 128 (2006) 11372-11373. |
| [9] |
Y. Li, K.J. Jardine, R. Tan, D. Song, V.M. Dong, Angew. Chem. Int. Ed. 48 (2009) 9690-9692. DOI:10.1002/anie.200905478 |
| [10] |
(a) Z. Chen, M. Zeng, J. Yuan, Q. Yang, Y. Peng, Org. Lett. 14 (2012) 3588-3591; (b) Z. Chen, X. Jia, C. Ye, G. Qiu, J. Wu, Chem. Asian J. 9 (2014) 126-130; (c) J. Tan, Z. Wang, J. Yuan, Y. Peng, Z. Chen, Adv. Synth. Catal. 361 (2019) 1295-1300; (d) H. Zhu, Z. Chen, Org. Lett. 18 (2016) 488-491; (e) Z. Chen, X. Jia, J. Huang, J. Yuan, J. Org. Chem. 79 (2014) 10674-10681. |
| [11] |
K. Strom, A. Impastato, K. Moy, A. Landreth, J. Snyder, Org. Lett. 17 (2015) 2126-2129.
|
| [12] |
(a) K. Albert, G. Schlotterbeck, U. Braumann, et al., Angew. Chem. Int. Ed. 34 (1995) 1014-1016; (b) B. Vaz, R. Alvarez, A.R. de Lera, J. Org. Chem. 67 (2002) 5040-5043. |
| [13] |
(a) J. Li, Y. Liang, Y. Xie, J. Org. Chem. 69 (2004) 8125-8127; (b) Y. Wen, L. Huang, H. Jiang, H. Chen, J. Org. Chem. 77 (2012) 2029-2034. |
| [14] |
S. Sarez-Pantiga, D. Palomas, E. Rubio, J. Gonzalez, Angew. Chem. Int. Ed. 48 (2009) 7857-7861. DOI:10.1002/anie.200902989 |
| [15] |
(a) G. Zhu, Z. Zhang, J. Org. Chem. 70 (2005) 3339-3341; (b) H. Jiang, C. Qiao, W. Liu, Chem. Eur. J. 16 (2010) 10968-10970; (c) S. Tang, Q. Yu, P. Peng, et al., Org. Lett. 9 (2007) 3413-3416; (d) D. Chen, X. Chen, Z. Lu, et al., Adv. Synth. Catal. 353 (2011) 1474-1478; (e) J. Li, S. Tang, Y. Xie, J. Org. Chem. 70 (2005) 477-479. |
| [16] |
(a) L. Bai, Y. Yuan, J. Liu, et al., Angew. Chem. Int. Ed. 55 (2016) 6946-6950; (b) H. Yoon, M. Rçlz, F. Landau, M. Lautens, Angew. Chem. Int. Ed. 56 (2017) 10920-10923; (c) J. Wallbaum, R. Neufeld, D. Stalke, D.B. Werz, Angew. Chem. Int. Ed. 52 (2013) 13243-13246; (d) B. Monks, S. Cook, J. Am. Chem. Soc. 134 (2012) 15297-15300; (e) S. Blouin, R. Pertschi, A. Schoenfelder, J. Suffert, G. Blond, Adv. Synth. Catal. 360 (2018) 2166-2171; (f) S. Blouin, V. Gandon, G. Blond, J. Suffert, Angew. Chem. Int. Ed. 55 (2016) 7208-7211; (g) J. Petrignet, A. Boudhar, G. Blond, J. Suffert, Angew. Chem. Int. Ed. 50 (2011) 3285-3289. |
| [17] |
(a) A. Reding, P. Jones, D.B. Werz, Angew. Chem. Int. Ed. 57 (2018) 10610-10614; (b) M. Pawliczek, T. Schneider, C. Maass, D. Stalke, D.B. Werz, Angew. Chem. Int. Ed. 54 (2015) 4119-4123; (c) T. Sperger, C. Le, M. Lautens, F. Schoenebeck, Chem. Sci. 8 (2017) 2914-2922; (d) C. Le, P. Menzies, D. Petrone, M. Lautens, Angew. Chem. Int. Ed. 54 (2015) 254-257. |
| [18] |
Z. Liu, M. Zhang, X. Wang, J. Am. Chem. Soc. 142 (2020) 581-588. DOI:10.1021/jacs.9b11909 |