 2021, Vol. 32
2021, Vol. 32 


Among various sulfur(VI) halides, sulfonyl fluorides have attracted increasing research attention in recent years due to their unique properties and widespread applications. In contrast with analogous sulfonyl chlorides, sulfonyl fluorides are incredibly stable and compatible with harsh reaction conditions, yet exhibiting excellent electrophilic reactivity towards O- and N-nucleophiles in a particular activating environment [1]. Although this unique reactivity-stability balance of organosulfur fluorides were recognized as early as in the 1920s [2], the more recent work of Sharpless and co-workers introduced the concept of sulfur(VI) fluoride exchange (SuFEx) and revived it from "old-school chemistry" to a new generation of click reactions [3]. SuFEx is emerging as a reliable tool for rapid assembly of functional molecules, and has quickly found extensive applications ranging from organic synthesis and polymer science to chemical biology and drug discovery (Fig. 1).

|
Download:
|
| Fig. 1. Representative sulfonyl fluorides with significant applications. | |
{kind=link}
In organic synthesis, sulfonyl fluorides are long regarded as versatile precursors instead of sulfonyl chlorides for the synthesis of sulfonyl-containing compounds including sulfonamides, sulfonate esters and sulfones [4]. Moreover, sulfonyl fluorides have been utilized as a new class of selective fluorinating reagents for deoxy-fluorination [5] and 18F radiolabelling [6]. More recently, fluorosulfuryl azide (FSO2N3) has been demonstrated as an excellent diazotransfer reagent for the synthesis of azides from primary amines [7].
In materials science, sulfonyl fluorides have been utilized for the synthesis of polymers bearing unique -SO2- linkages, which is a significant complement to classic synthetic polymers featuring carbon-carbon or carbon-heteroatom linkages. Moreover, owing to the orthogonal reactivity of SuFExable groups, -SO2F moieties have been used as reactive handles in side chains, which can be smoothly pre-assembled onto monomers, for post-polymerization modification of polymers [8].
In the biological discipline, sulfonyl fluorides have been used as privileged covalent probes and inhibitors in chemical biology and drug discovery [9]. They are demonstrated to be able to covalently modify various amino acid residues of enzyme binding sites, including context-specific serine, threonine, lysine, tyrosine, cysteine, and histidine residues. In contrast with the widely adopted copper-catalyzed alkyne-azide cycloadditions (CuAAC) reactions, SuFEx reactions have unique characteristics [1a]. Firstly, SuFEx reactions are generally performed under metal-free conditions, which have particular significance regarding the application of click chemistry in biological systems. Secondly, SuFEx reactions generate more diverse connections with various nucleophiles, ranging from silyl ethers, alcohols, amines to carbon nucleophiles, to create diverse functional molecules through S-F exchange. Thirdly, in contrast to the defined terminal alkyne and azide functional groups that require multiple steps to be installed into substrates for CuAAC reactions, the SuFExable handles are generally more flexible and easier to be introduced by uniting common native functional groups through discrete connective hubs, such as SO2F2 and SOF4. These features of SuFEx reactions will enable it with broader utilization in chemical biology and drug discovery.
Given the aforementioned power of sulfonyl fluorides in extensive fields, the development of efficient approaches for obtaining sulfonyl fluorides is undoubtedly in high demand and has become a hot topic in organic chemistry. In this review, we aim to give a timely summary on synthetic methods of sulfonyl fluorides and provide our viewpoints on existing challenges and further development directions. Meanwhile, this review is organized according to synthetic methods of different kinds of RSO2F, which include aromatic, aliphatic, alkenyl, and alkynyl sulfonyl fluorides.
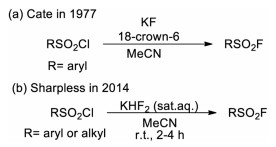
2. Synthesis of arenesulfonyl fluorides (Ar-SO2F) 2.1. From (hetero)aromatic sulfur compoundsThe most conventional approach for the synthesis of arenesulfonyl fluorides involves the nucleophilic fluorination of the corresponding sulfonyl chlorides in the presence of a suitable fluoride source, and the pioneering work was reported in 1931 by Davies and Dick [10]. By boiling the mixture of aromatic or aliphatic sulfonyl chlorides with aqueous potassium fluoride solution, the corresponding sulfonyl fluorides were readily prepared. In 1977, Cate and co-workers developed an efficient "naked fluoride" method (using KF and 18-crown-6 in dry acetonitrile) for the preparation of sulfonyl fluorides (Scheme 1a) [11]. However, the occurrence of side reactions and the hydrolysis of alkyl sulfonyl fluorides under this condition may overshadow the advantages of this method [12]. To address this challenge, Sharpless and co-workers developed an improved method using potassium bifluoride as the alternative fluoride source (Scheme 1b) [3]. With this method, alkyl and aryl sulfonyl chlorides can be efficiently transformed into corresponding sulfonyl fluorides in high yield under mild condition without hydrolysis of the sulfonyl fluoride products. It was proposed that the solvation and hydrogen bond play an important role at the water/organic interface for increasing the nucleophilicity of the fluoride ion toward fluoride-chloride exchange.

|
Download:
|
| Scheme 1. Synthesis of sulfonyl fluorides via F-Cl exchange. | |
{kind=link}
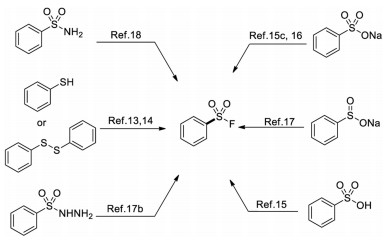
In view of the highly sensitive reactivity and poor accessibility of sulfonyl chlorides, alternative methods for synthesizing sulfonyl fluorides have been developed through employing a more diverse set of starting materials. Aromatic sulfur compounds such as thiols [13], disulfides [14], sulfonic acid [15], sodium sulfonates [15c, 16], sulfinates [17], sulfonyl hydrazides [17b] and sulfonamides [18] have been demonstrated to be alternative synthetic precursors for the corresponding sulfonyl fluorides (Scheme 2).

|
Download:
|
| Scheme 2. Synthesis of sulfonyl fluorides from various aromatic sulfur compounds. | |
{kind=link}
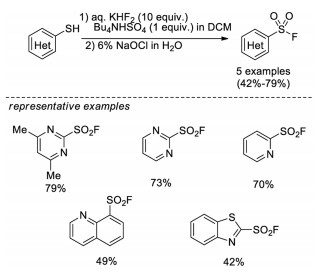
In 2006, Wright and Hallstrom developed an efficient method to synthesize heterocyclic sulfonyl fluorides from thiols [13a]. This process involves an oxidative chlorination step by using aqueous sodium hypochlorite to afford the sulfonyl chloride intermediate in situ, followed by fluoride-chloride exchange to form sulfonyl fluorides. This reaction uses readily available reagents, and avoids the use of chlorine gas. However, only heteroaromatic thiols were demonstrated in this methodology (Scheme 3).

|
Download:
|
| Scheme 3. Synthesis of heterocyclic sulfonyl fluorides from thiols. | |
{kind=link}
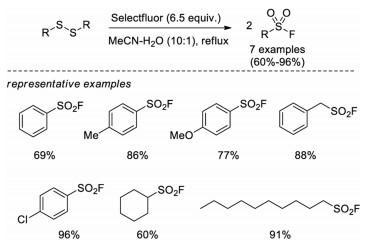
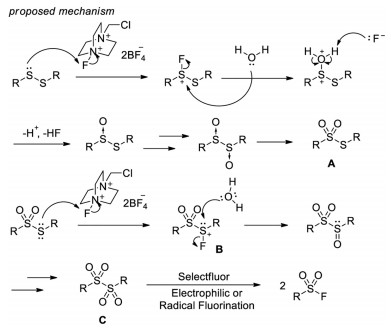
In 2011, Kirihara and co-workers introduced a method to transform disulfides into sulfonyl fluorides with Selectfluor as both oxidant and fluorine source (Scheme 4) [14]. In this system, aromatic, benzylic and alkyl disulfides can be converted to corresponding sulfonyl fluorides in good yields. Control experiments demonstrated that a thiosulfate intermediate A was generated in this process and the loading of Selectfluor was critical. Although the exact mechanism remains unclear, the authors proposed that electrophilic fluorination of the sulfur atom of the thiosulfate A to form a fluorinated sulfonium salt B, followed by addition of water to afford a dithiosulfinate C, which subsequently undergoes radical or electrophilic fluorination to give the final product sulfonyl fluoride (Scheme 5).

|
Download:
|
| Scheme 4. Synthesis of sulfonyl fluorides from thiols or disulfides. | |
{kind=link}

|
Download:
|
| Scheme 5. Synthesis of sulfonyl fluorides from thiols or disulfides using excess Selectfluor as oxidant. | |
{kind=link}
In 2019, Noël and co-workers reported an efficient electrochemical oxidative approach to afford sulfonyl fluorides from widely available thiols and disulfides with KF as a cheap fluorine source [13b]. This green and mild protocol doesnot require additional oxidants or catalysts, and displays a broad substrate scope, including aryl, heteroaryl, benzyl, and alkyl thiols or disulfides (Scheme 6).

|
Download:
|
| Scheme 6. Synthesis of sulfonyl fluorides via electrochemical oxidative of thiols or disulfides. | |
{kind=link}
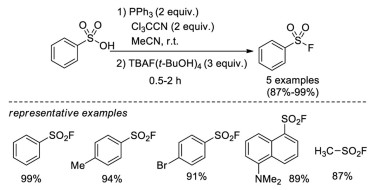
In 2010, Jang and co-workers developed a two-step one-pot procedure for the synthesis of sulfonyl fluorides from sulfonyl acids [15b]. The reaction used a combination of trichloroacetonitrile and triphenylphosphine to generate sulfonyl chloride in situ, which then was transformed into the corresponding sulfonyl fluorides by treatment with TBAF (t-BuOH)4. Several examples were explored in this approach, with high yield in short reaction times (Scheme 7).

|
Download:
|
| Scheme 7. Synthesis of sulfonyl fluorides from sulfonic acids. | |
{kind=link}
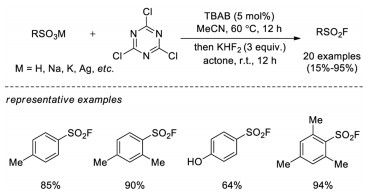
Recently, Qin and co-workers developed an efficient and mild method for the preparation of sulfonyl fluorides from sulfonates or sulfonic acids through chlorination with cyanuric chloride and followed by chlorine-fluorine exchange (Scheme 8) [15c].

|
Download:
|
| Scheme 8. Synthesis of sulfonyl fluorides from sulfonates or sulfonic acids. | |
{kind=link}
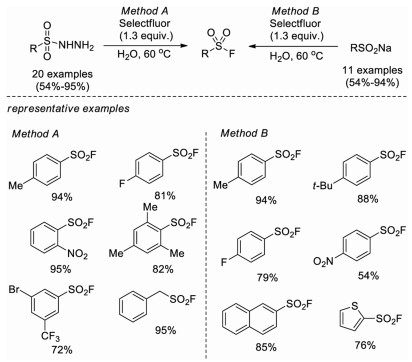
In 2016, Wang and co-workers disclosed a catalyst-free fluorination of sulfonyl hydrazides for the synthesis of sulfonyl fluorides using Selectfluor as the fluorine source [17b]. This approach shows a broad substrate scope and does not require any metal catalyst and other additive. A wide range of aryl and alkyl sulfonyl fluorides were obtained under mild conditions in water. On the basis of several control experiments, it was proposed that sulfonyl hydrazide could liberate nitrogen and form sulfonyl radical in the presence of Selectfluor, then the sulfonyl radical rapidly captures fluorine to generate sulfonyl fluoride. Notably, sodium benzenesulfinate could also react smoothly with Selectfluor to give benzenesulfonyl fluoride under the same condition and even not affected by the radical inhibitor TEMPO (2, 2, 6, 6-tetramethyl-1-piperidinyloxy). Several sodium arylsulfinates were explored effectively under this condition (Scheme 9).

|
Download:
|
| Scheme 9. Synthesis of sulfonyl fluorides from sulfonyl hydrazides or sodium benzenesulfinate in water. | |
{kind=link}
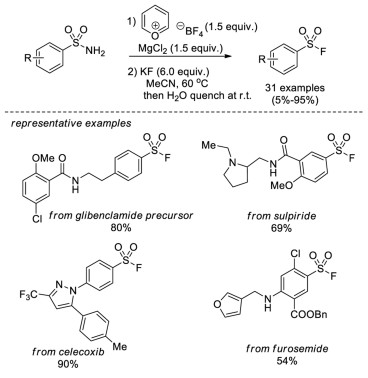
In 2020, Cornella and co-workers reported a new protocol for the synthesis of sulfonyl fluorides from primary sulfonamides [18], which are conventionally regarded as relatively unreactive compounds in organic synthesis. This method was in line with their previous work for the deaminative chlorination by means of a pyrylium salt (Pyry-BF4) and MgCl2 [19]. After subsequent in situ fluoride-chloride exchange with KF, a variety of sulfonamides were efficiently converted into sulfonyl fluorides. Notably, the mild reaction conditions and the excellent functional group tolerance allows for the late-stage formation of sulfonyl fluorides from complex and densely functionalized primary sulfonamides (Scheme 10).

|
Download:
|
| Scheme 10. Synthesis of sulfonyl fluorides from sulfonamides. | |
{kind=link}
2.2. From (hetero)aryl halides
Apart from using (hetero)aromatic sulfur compounds, a more diverse set of starting materials have also been explored for the synthesis of sulfonyl fluorides.
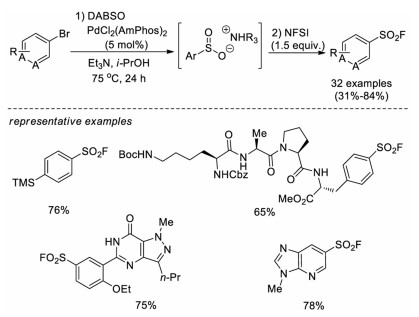
In 2017, Willis and co-workers reported a pioneer work for the synthesis of sulfonyl fluorides from aryl or heteroaryl bromides [20]. This approach was the first one that used non-sulfur compounds as starting materials. The two-step, one-pot method involved the initial palladium-catalyzed sulfonylation of aryl bromides using DABSO (1, 4-diazabicyclo[2.2.2]octane bis(sulfur dioxide)) as an SO2 source to generate an ammonium sulfinate intermediate, followed by in situ electrophilic fluorination of the sulfinate intermediate with NFSI (N-fluorobenzenesulfonimide). A variety of aryl and heteroaryl sulfonyl fluorides were afforded in moderate to good yields. Notably, this strategy was efficient for the late-stage introduction of fluorosulfonyl moiety into pharmaceutical intermediates. Moreover, a series of aryl, benzyl, and alkyl Grignard reagents can also be converted to corresponding sulfonyl fluorides by successive treatment with DABSO and NFSI (Scheme 11).

|
Download:
|
| Scheme 11. Pd-catalyzed synthesis of sulfonyl fluorides from aryl bromides. | |
{kind=link}
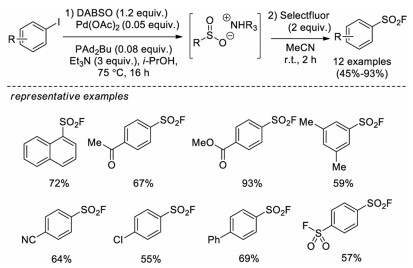
Shortly thereafter, Ball and co-workers developed a similar approach to synthesize aryl sulfonyl fluorides from aryl iodides catalyzed by Pd(OAc)2 with Selectfluor as the fluorine source (Scheme 12) [21].

|
Download:
|
| Scheme 12. Pd-catalyzed synthesis of sulfonyl fluorides from aryl iodides. | |
{kind=link}
In 2019, Kim and co-workers developed a transition-metal-free synthesis of 2-aminoarenesulfonyl fluoride derivatives from aryne precursors, secondary amines and sulfuryl fluoride (SO2F2) [22]. A series of 2-dialkyl-, 2-alkylaryl-, or 2-diarylamino-substituted aryl sulfonyl fluoride derivatives were obtained in good to excellent yields in this system. The authors suggested that zwitterionic intermediate D was formed by the nucleophilic attack of amines to the in-situ generated arynes, which then could capture SO2F2 to form aryl sulfonyl fluorides. Furthermore, it was envisioned that a strong hydrogen bond between the ammonium salt and SO2F2 contributed to the high electrophilicity of SO2F2 and high regioselectivity for nucleophilic attack (Scheme 13).

|
Download:
|
| Scheme 13. Synthesis of sulfonyl fluorides by sulfuryl fluoride incorporation to arynes. | |
{kind=link}
In 2019, Sammis, Ball and co-workers reported a one-pot fluorosulfonylation reaction using Grignard reagents and sulfuryl fluoride (SO2F2) [23]. A series of aryl, alkyl and heteroaryl Grignard reagents were converted to desired sulfonyl fluorides in a solution of sulfuryl fluoride at ambient temperature. However, the substrate scope was relatively limited, and was not efficient for strongly electron-withdrawing substituted phenyl derivatives (Scheme 14).

|
Download:
|
| Scheme 14. Synthesis of sulfonyl fluorides from Grignard reagents using sulfuryl fluoride. | |
{kind=link}
2.3. From aromatic amines
The aromatic amino group is widely found in numerous pharmaceutical drugs and bioactive compounds. Although various synthetic conversions of aryldiazonium salts have been successfully developed, there are no successful examples of their fluorosulfonylation reaction. Until recently, Liu and Chen reported a copper-catalyzed fluorosulfonylation reaction of aryldiazonium salts in the presence of catalytic amounts of CuCl2 and 6, 6′-dimethyl-2, 2′-dipyridyl at room temperature [24]. A wide range of arylsulfonyl fluorides were obtained using DABSO as the sulfonyl source in combination with KHF2 as the fluorine source. Two possible mechanistic pathways were proposed according to different aryldiazonium salts. The common step is the generation of arylsulfonyl radical [ArSO2·]via trapping SO2 by the aryl radical, which is generated from the corresponding arenediazonium salt via a single-electron transfer with Cu(I) species or DABSO. For arenediazonium salts bearing electron-donating group, the arenesulfonyl radical [ArSO2·] abstracts chlorine from Cu(II) species to form arylsulfonyl chloride [ArSO2Cl] and regenerates Cu(I) species, then fluoride-chloride exchange results the arylsulfonyl fluoride (Scheme 15, path A). For arenediazonium salts bearing electron-withdrawing group, [ArSO2·] reacts with fluorine anion to form a radical anion intermediate [ArSO2F·−], and its back-electron transfer with aryldiazonium salt results to arylsulfonyl fluoride and regenerates aryl radical (Scheme 15, path B).

|
Download:
|
| Scheme 15. Copper-catalyzed fluorosulfonylation of arenediazonium salts. | |
{kind=link}
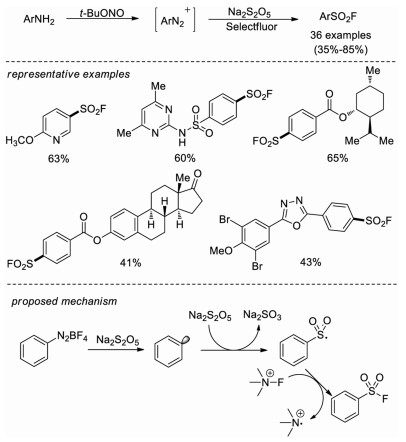
Almost at the same time, Weng and co-workers developed a copper-free Sandmeyer-type fluorosulfonylation reaction [25]. Aryldiazonium salts were converted to arylsulfonyl fluorides utilizing sodium metabisulfite (Na2S2O5) and Selectfluor as the sulfur dioxide surrogate and fluorine sources, respectively, without any other additives. This method tolerates a wide range of substrates, including substrates with sensitive functional groups (-COOH, -CN, -Br, etc.) and complex molecular architectures are tolerated. The two-step, one-pot synthesis of sulfonyl fluorides from aromatic amines and late-stage fluorosulfonylation of natural products or pharmaceuticals were also realized in this methodology. Mechanistic studies showed that both aryl radical and arylsulfonyl radical were generated in this system, and subsequent radical fluorination afforded the final sulfonyl fluoride product. It was also proposed the sodium metabisulfite might act as both the reductant and the sulfur dioxide surrogate (Scheme 16).

|
Download:
|
| Scheme 16. Copper-free synthesis of sulfonyl fluorides from aryldiazonium salts. | |
{kind=link}
Shortly thereafter, two similar works focusing on the construction of aromatic sulfonyl fluorides from aryldiazonium salts were developed by the groups of Liu and Qing, respectively (Scheme 17) [26].

|
Download:
|
| Scheme 17. Liu's and Qing's work for the fluorosulfonylation of arenediazonium salts. | |
{kind=link}
3. Synthesis of alkenylsulfonyl fluorides (C(sp2)-SO2F) 3.1. ESF as the connector
Ethenesulfonyl fluoride (ESF), which is a useful and readily available SO2F-containing reagent, was first described by Hedrick in 1953 and well-studied by Hyatt in 1979 [27]. However, this small, highly connective module has been overlooked in chemistry research until the introduction of SuFEx chemistry in 2014. Over the past few years, the application of ESF has received increasing attention in the synthesis of sulfonyl fluorides [28]. ESF has proven to be a versatile reagent in various transformations, including Michael addition, radical addition, and cycloaddition [3, 29].
In 1979, Hyatt and co-workers reported a three-step synthesis of ESF from sodium 2-hydroxyethanesulfonate in 55% overall yield (Scheme 18a) [27b]. In 2016, Sharpless and co-workers developed an improved "on-water" approach for the kilogram-scale preparation of ESF in 98% overall yield (Scheme 18b) [30]. The first step involves the chloride-fluoride exchange of 2-chloroethenesulfonyl chloride with potassium bifluoride. A subsequent dehydrochlorination of 2-chloroethanesulfonyl fluoride using magnesium oxide affords the desired ESF. This efficient method helped the chemical community to access sulfonyl fluorides more easily.

|
Download:
|
| Scheme 18. General method for the synthesis of ethenesulfonyl fluoride. | |
{kind=link}
3.1.1. Heck-type cross-coupling reactions with ESF
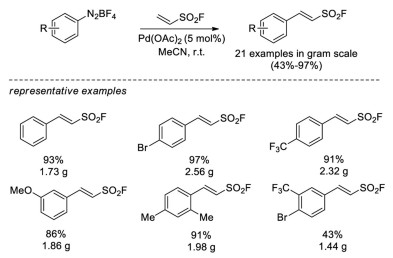
In 2016, Wu and Sharpless reported the Heck-Matsuda cross-coupling between the readily prepared aryldiazonium tetrafluoroborate salts and ESF [31]. Various 2-arylethenesulfonyl fluorides were obtained in 43%–97% yield catalyzed by Pd(OAc)2 catalyst. They also demonstrated that the 2-arylethenesulfonyl fluorides could be a flexible bis-electrophiles for SuFEx click chemistry, in which either the C=C double bond or the SO2F group can be selectively attacked by nucleophiles under certain conditions (Scheme 19).

|
Download:
|
| Scheme 19. Pd-catalyzed Heck-Matsuda cross-coupling of aryldiazonium tetrafluoroborates and ESF. | |
{kind=link}
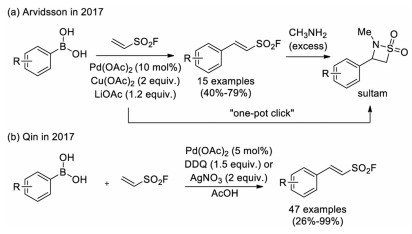
Shortly after, Arvidsson and co-workers reported an oxidative Boron-Heck coupling between ESF and commercially available aryl boronic acids for the preparation of β-aryl/heteroaryl ethenesulfonyl fluorides at room temperature (Scheme 20a) [32]. In addition to the Pd(OAc)2 catalyst, stoichiometric amount of Cu(OAc)2 and LiOAc were employed as the oxidant and base, respectively. Moreover, one-pot synthesis of β-sultams via the subsequent addition of excess amine was also realized. In 2017, Qin and co-workers reported a similar oxidative Heck reaction using 2, 3-dichloro-5, 6-dicyano-1, 4-benzoquinone (DDQ) or AgNO3 as the oxidant in acetic acid at 80℃ (Scheme 20b) [33]. This method presented broader substrate scope and gave higher yields. The acidic condition in this approach was also an appropriate complement with Arvidsson's basic condition approach.

|
Download:
|
| Scheme 20. Oxidative Heck coupling of arylboronic acids and ESF. | |
{kind=link}
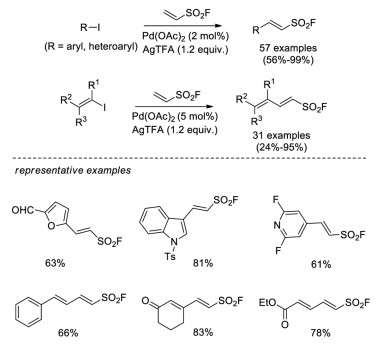
In 2017, Qin and Sharpless developed another Pd-catalyzed Heck coupling reaction of ESF with various aryl, heteroaryl and alkenyl iodides [34]. This method relied on Pd(OAc)2 catalyst with stoichiometric AgTFA as the halide scavenger, affording 88 structurally diverse vinyl sulfonyl fluorides in up to 99% yields, including unprecedented β-heteroaryl-substituted vinylsulfonyl fluorides and dienylsulfonyl fluorides (Scheme 21).

|
Download:
|
| Scheme 21. Heck coupling of organic iodides and ESF. | |
{kind=link}
3.1.2. Carbon-hydrogen functionalization with ESF
Apart from these Heck cross-coupling reactions mentioned above, direct C–H activation represents an alternative strategy for the introduction of ESF into arenes.
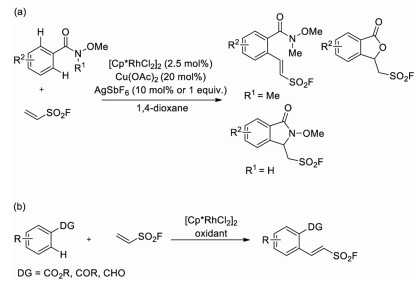
In 2018, Qin and co-workers reported the first Rh(III)-catalyzed C–H activation strategy for the synthesis of 2-arylethenesulfonyl fluorides via the oxidative coupling of N-methoxybenzamides with ESF (Scheme 22a) [35], where the amide functional group was employed as an ortho-directing group. This protocol featured an exclusive E-stereoselectivity of final products and a monoselective ortho-functionalization of sp2 C-H bonds of the aryl rings.

|
Download:
|
| Scheme 22. Rh-catalyzed directed C-H activation for the insertion of ESF moiety. | |
{kind=link}
Later, the same group demonstrated that arylcarboxylic esters, ketones and aldehydes could also be employed as directing groups in Rh-catalyzed functionalization of ortho C–H bonds with ESF (Scheme 22b) [36].
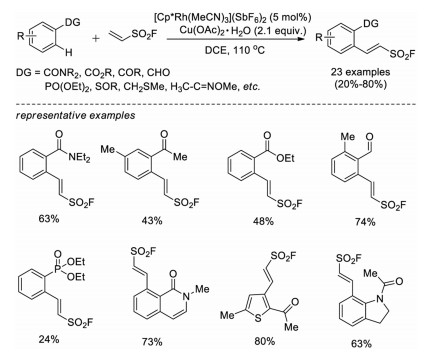
In 2019, Huestis and co-workers reported a similar rhodium(III)-catalyzed approach for C–H fluorosulfonylvinylation of various arenes bearing diverse directing groups, including amides, phosphonates, sulfoxides, sulfides, oximes, and carbonyl groups [37]. Although low yields were obtained in some cases, the scope of applicable directing groups has been greatly expanded (Scheme 23).

|
Download:
|
| Scheme 23. Rh-catalyzed fluorosulfonylvinylation of arenes with various directing groups. | |
{kind=link}
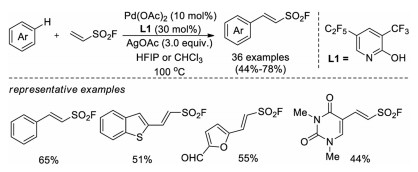
In 2019, Wang, Yu, and co-workers reported a Pd-catalyzed non-directed C-H functionalization of arenes with ESF to prepare β-aryl vinyl sulfonyl fluorides [38]. An electron-deficient 2-pyridone ligand is crucial for this reaction with arenes as the limiting reagent. This protocol tolerates a variety of functional groups, and late-stage fluorosulfonylvinylation of various pharmaceuticals and their further click manipulations were also demonstrated (Scheme 24).

|
Download:
|
| Scheme 24. Pd-catalyzed, non-directed C-H activation of arenes with ESF. | |
{kind=link}
3.1.3. Cycloaddition of ESF
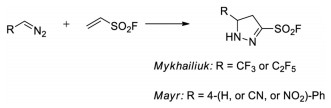
In 2014, Mykhailiuk and co-workers reported a [3+2] cycloaddition reaction between diazomethanes and electro-deficient alkenes or alkynes, including ESF [39]. In 2018, Mayr and co-workers also reported several examples of [3+2] cycloaddition reaction with ESF to give 3-sulfonyl fluoride substituted 5-arylpyrazolines (Scheme 25) [40]

|
Download:
|
| Scheme 25. [3+2] cycloaddition with ESF. | |
{kind=link}
3.2. 1-Br-ESF as the connector 3.2.1. Cycloaddition of 1-Br-ESF
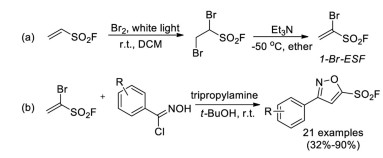
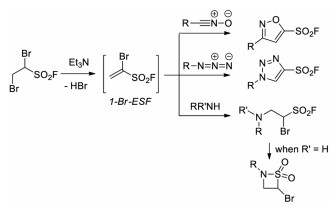
In 2018, Qin and co-workers developed a novel SuFEx clickable reagent 1-bromoethene-1-sulfonyl fluoride (1-Br-ESF) (Scheme 26a) [41]. Owing to the additional reactive bromide moiety, 1-Br-ESF would have great potential to enrich the SuFEx click chemistry toolbox. 1-Br-ESF was readily prepared from ESF on a 40-gram scale. Meanwhile, the 1, 3-dipolar cycloaddition between 1-Br-ESF and N-hydroxybenzimidoyl chlorides was realized to for the synthesis of a series of 5-sulfonylfluoro isoxazoles, which demonstrated the potential application of this reagent (Scheme 26b).

|
Download:
|
| Scheme 26. The synthesis and application of 1-Br-ESF. | |
{kind=link}
Soon after, the groups of Moses and Fokin demonstrated that the reactive 1-Br-ESF could undergo regiospecific 1, 3-dipolar cycloaddition reactions with a number of organic azides and N-hydroxyimidoyl chlorides to give the corresponding SO2F- substituted 5-membered aromatic heterocycles in good to excellent yields [42]. Notably, 1-Br-ESF could be in situ generated from 1, 2-dibromoethane-1-sulfonyl fluoride (DESF) via dehydrobromination [42a]. Moreover, as a powerful Michael acceptor, 1-Br-ESF could also react with secondary or primary amines to generate 1, 4-addition products and 4-Br-β-sultams, respectively. SuFEx reaction for post-synthetic modifications of the SO2F-functionalized aromatic heterocycles was also demonstrated to convenient access to various sulfonates, sulfonamides, and sulfonic acid derivatives (Scheme 27).

|
Download:
|
| Scheme 27. The in-situ formation of 1-Br-ESF and its application. | |
{kind=link}
3.2.2. Suzuki-type cross-coupling reactions with 1-Br-ESF
In 2019, Qin and co-workers developed a Suzuki coupling of arylboronic acids with 1-Br-ESF to give α-aryl ethenesulfonyl fluorides, while no competitive Heck-type products were detected in this system [43]. This method provides a vast of alkenylsulfonyl fluorides, which could be used as building blocks for further transformation (Scheme 28).

|
Download:
|
| Scheme 28. Suzuki coupling of arylboronic acids and 1-Br-ESF. | |
{kind=link}
3.3. BTESF as the connector
In 2020, Qin and co-workers developed a newly clickable connective hub, 1-bromo-2-triazolethane-1-sulfonyl fluoride (BTESF), which can be prepared from ESF, and demonstrated its utility in the construction of a variety of enaminyl sulfonyl fluorides (N-ESF) [44]. Late-stage fluorosulfonylation of pharmaceuticals were also realized in this methodology and antimicrobial experiments of these vinyl sulfonyl fluoride functionalized drugs indicated the antibacterial activities against MRSA (methicillin resistant Staphylococcus aureus) and SA (Staphylococcus aureus). The transformation underwent a sequential elimination/Michael addition/elimination process (Scheme 29).

|
Download:
|
| Scheme 29. Construction of enaminyl sulfonyl fluorides using 1-bromo-2-triazolethane-1-sulfonyl fluoride (BTESF) as a SuFEx-enabling reagent. | |
{kind=link}
3.4. Other methods
In 2019, Willis and co-workers reported a palladium-catalyzed synthesis of cyclic alkenylsulfonyl fluorides from the corresponding alkenyl triflates [45]. In these reactions, the alkenyl triflates substrates react with DABSO via the Pd-catalyzed sulfur dioxide insertion, followed by electrophilic fluorination using NFSI to afford the products. A series of cyclic alkenylsulfonyl fluorides bearing various functional groups were obtained in moderate to good yields. The authors also demonstrated the practicality of these new reagents via diverse orthogonal derivatization reactions, including nucleophilic substitution at sulfur, conjugate addition, catalytic hydrogenation, and N-functionalization, which is a quite useful addition to the SuFEx click chemistry toolbox (Scheme 30).

|
Download:
|
| Scheme 30. Pd-catalyzed synthesis of cyclic alkenylsulfonyl fluorides via sulfur dioxide insertion. | |
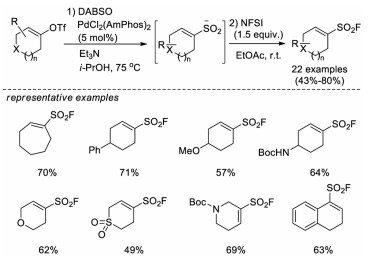{kind=link}
In 2020, Liao and co-workers developed a novel radical fluorosulfonylation reaction with a fluorosulfonyl radical (FSO2·) generated from FSO2Cl under photoredox conditions [46]. A wide range of alkenes are smoothly converted to alkenyl sulfonyl fluorides, including structures that would otherwise be challenging to synthesize via the known cross coupling methods with ESF, such as β-alkyl alkenes, cyclic, di- and tri-substituted olefins, and some natural products or its derivatives. It is worth noting that this is the first fluorosulfonylation reaction with fluorosulfonyl radical (Scheme 31).

|
Download:
|
| Scheme 31. Radical fluorosulfonylation of olefins. | |
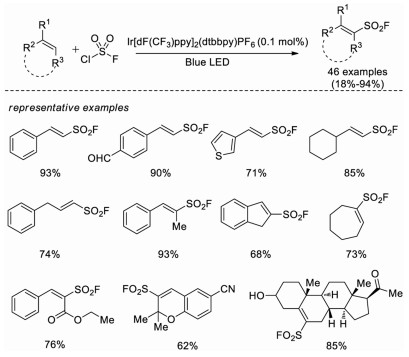{kind=link}
4. Synthesis of aliphatic sulfonyl fluorides (C(sp3)-SO2F)
In contrast to arylsulfonyl fluorides, the construction of aliphatic sulfonyl fluorides is relatively difficult and less reported. The typical method is the fluorine-chloride exchange of alkyl sulfonyl chlorides (Section 2). In addition, alkenylsulfonyl fluorides have also be used as a building block for the synthesis of aliphatic sulfonyl fluorides.
4.1. From alkyl halidesIn 2016, Shavnya and co-workers developed an efficient method for one-pot synthesis of aliphatic sulfonyl fluorides, sulfonamides, or sulfones from primary and secondary alkyl halides, using sodium hydroxymethylsulfinate (Rongalite) as the SO2 surrogate. A handful of sulfonyl fluorides were synthesized when using NFSI as the quenching electrophile for the last step (Scheme 32a) [47]. In 2018, the same group developed a new sulfonylating reagent (Rongacyl), which is a SO22− anion equivalent derived from rongalite. Several alkyl sulfonyl fluorides were afforded in a multi-step manner on the basis of alkylation of this sulfonylating reagent (Scheme 32b) [48].

|
Download:
|
| Scheme 32. Multistep synthesis of aliphatic sulfonyl fluorides from alkyl halides. | |
{kind=link}
4.2. From alkenylsulfonyl fluorides
The most powerful sulfur(VI) hub for the synthesis of alkyl sulfonyl fluorides is ESF, a highly reactive Michael acceptor, dienophile and 1, 3-dipolarphile.
4.2.1. Conjugate addition reactions of ESFAs early as in 1979, Hyatt and co-workers investigated the Michael reaction of ESF with amines, thiols, as well as active methylene nucleophiles, affording a wide range of aliphatic sulfonyl fluorides in excellent yields [27b].
In 2014, Sharpless and co-workers smoothly extended the Michael reaction of ESF with various nucleophiles, which included active amines, amine-containing zwitterions, sulfonamides, alcohols, and 1, 3-dicarbonyl compounds [3]. It is impressive that purification is rarely required for most ESF-amine conjugate additions (Scheme 33).

|
Download:
|
| Scheme 33. Conjugate addition of carbon, oxygen, and nitrogen nucleophiles to ESF. | |
{kind=link}
In 2019, Leung and co-workers reported the first Pd-catalyzed enantioselective hydrophosphination reaction of β-arylethenesulfonyl fluorides [49]. A series of chiral sulfonyl fluoride-derived phosphine were obtained in excellent yields and with up to 93% ee value (Scheme 34).

|
Download:
|
| Scheme 34. Pd-catalyzed enantioselective hydrophosphination reaction of β-arylethenensulfonyl fluorides. | |
{kind=link}
In 2019, Qin and co-workers achieved a Rh-catalyzed 1, 4-conjugate addition of 1, 3-dienylsulfonyl fluorides with (hetero)aryl boronic acids (Scheme 35a) [50]. A variety of (E)-2-aryl-4-(aryl or alkyl)but-3-ene-1-sulfonyl fluorides were obtained in high yields with exclusive regioselectivities. Besides, under the control of chiral ligand, 12 examples of chiral β-aryl sulfonyl fluorides were synthesized with high yields and enantioselectivities in the presence of a C1-sysmmetric bicyclo[2.2.2]octa-2, 5-diene ligand L2. In the same year, this group realized the Rh-catalyzed asymmetric conjugate addition of arylboronic acids with 2-arylethenesulfonyl fluorides (Scheme 35b) [51]. A variety of novel sulfonyl fluorides bearing chiral gem-diarylmethane moiety were obtained with excellent enantioselectivities (up to 99.8% ee).

|
Download:
|
| Scheme 35. Rh-catalyzed regioselective and enantioselective 1, 4-conjugate addition to 1, 3-dienylsulfonyl fluorides with arylboronic acids. | |
{kind=link}
In 2019, Yan and co-workers achieved an organocatalytic enantioselective Michael addition of 3-amido-2-oxindoles to ESF [52]. A series of chiral sulfonyl fluorides with 3, 3-disubstituted 2-oxindole scaffolds were prepared with excellent yields and enantioselectivities. It was proposed that the quinine-derived squaramide E could simultaneously activate the two substrates via hydrogen-bonding interaction. Furthermore, the products were smoothly transformed to chiral spirocyclic oxindole sultams, sulfamide, and sulfonate without loss of the optical purity (Scheme 36a).

|
Download:
|
| Scheme 36. Asymmetric conjugate addition to ESF. | |
{kind=link}
In 2021, the same group developed an enantioselective Michael addition of N-2, 2, 2-trifluoroethylisatin ketimines to ESF, providing a series of chiral oxindoles bearing C(sp3)-SO2F groups [53]. Excellent yields and enantioselectivities were obtained with the same quinine-derived squaramide catalyst E (Scheme 36b).
In 2021, Qin, Tang and co-workers developed a copper-catalyzed, three component reaction for the construction of indolizine-containing aliphatic sulfonyl fluorides under mild conditions [54]. Quinolines, isoquinolines and pyridines are tolerated in this reaction. A plausible mechanism was postulated Firstly, a key precursor 1, 3-dipole F was formed under copper catalysis, subsequently underwent cycloaddition of F with ESF to generate the intermediate G. Then, G underwent deprotonation and Michael addition with another molecule of ESF to provide intermediate H. Finally, under the driving force of aromatization/conjugation together with the promotion of basic conditions, intermediate H was transformed into the final product in multiple steps (Scheme 37).

|
Download:
|
| Scheme 37. Copper-catalyzed cycloaddition/Michael addition tandem reaction. | |
{kind=link}
4.2.2. Cycloaddition reactions of ESF
Owing to the electron-withdrawing property and good stability of -SO2F group, the groups of Daeniker and Hyatt used ESF as the dienophile in Diels-Alder reactions with cyclic dienes (Scheme 38a) [27b, 55]. In 1987, Zéroual and co-workers reported the [3+2] cycloaddition reaction of ESF or 1-Br-ESF with nitrones to give isoxazolidine-derived sulfonyl fluorides (Scheme 38b) [56]. In 2018, Mykhailiuk and co-workers developed an example of photocatalytic [2+2] cycloaddition reaction of ESF and N-benzylmaleimide (Scheme 38c) [57].

|
Download:
|
| Scheme 38. Cycloaddition reaction with ESF. | |
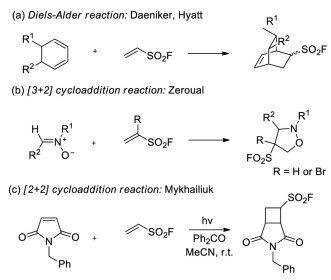{kind=link}
In 2018, Grygorenko and co-workers developed a [3+2] cycloaddition reaction between α, β-unsaturated sulfonyl fluorides and in-situ generated azomethyne ylides to form a series of pyrrolidine-3-sulfonyl fluorides [58]. Notably, the reaction can be scaled up to 25 g without obvious loss of yield (Scheme 39).

|
Download:
|
| Scheme 39. [3+2] cycloaddition reaction with α, β-unsaturated sulfonyl fluorides. | |
{kind=link}
In 2020, Qin and co-workers reported an efficient photocatalytic [2+2] cycloaddition reaction between pyridones or isoquinolones and ESF or 1-Br-ESF, providing a variety of unique cyclobutene-fused pyridinyl sulfonyl fluorides (Scheme 40) [59].

|
Download:
|
| Scheme 40. Photocatalytic [2+2] cycloaddition reaction between pyridones or isoquinolones and ESF or 1-Br-ESF. | |
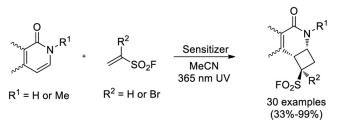{kind=link}
4.2.3. Radical addition reactions of ESF
In 2019, Liao and co-workers developed a visible-light-mediated decarboxylative fluorosulfonylethylation approach for the synthesis of aliphatic sulfonyl fluorides using ESF as the radical acceptor [60]. A variety of NHPI redox active esters derived from alkyl carboxylic acid feedstocks proceeded smoothly under photoredox catalytic conditions to give corresponding aliphatic sulfonyl fluorides in high yields. Further diversification of the SO2F-containing products was also demonstrated (Scheme 41).

|
Download:
|
| Scheme 41. Visible-light-mediated radical addition to ESF. | |
{kind=link}
In 2020, Qin and co-workers reported a visible light-induced reductive addition reaction of alkyl iodides with ESF, employing Hantzsch ester as a hydrogen source [61]. A broad range of 1°, 2° and 3° alkyl iodides were efficiently transformed to corresponding aliphatic sulfonyl fluorides, including derivative compounds from enzyme inhibitors, natural products and pharmaceuticals (Scheme 42).

|
Download:
|
| Scheme 42. Visible light-induced reductive addition of alkyl iodides to ESF. | |
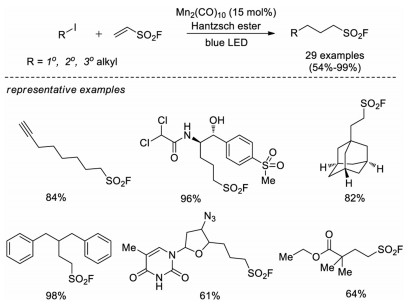{kind=link}
4.3. Difunctionalization of alkenes
In 2017, Liu, Chen and co-workers developed a novel intermolecular radical reaction of unactivated alkenes to realize the construction of C(sp3)-SO2F [62]. In this strategy, readily available Ag(O2CCF2SO2F) provides both trifluoromethyl and SO2 sources for addition across alkene, followed by fluorination with N-fluorobenzenesulfonimide (NFSI) to yield the final trifluoromethylfluorosulfonylation products. A variety of substrates with unactivated alkene were tolerated in this condition. According to control experiments, it was proposed Ag(O2CCF2SO2F) easily releases CO2 and SO2 molecules to form AgCF3 under reaction conditions. The AgCF3 species generates a CF3 radical which can add to the alkene affording a new alkyl radical. Alkyl radical captures SO2 to form sulfonyl radical, and later electrophilic fluorination results in 1, 2-difunctionalization products (Scheme 43).

|
Download:
|
| Scheme 43. Intermolecular radical trifluoromethylfluorosulfonylation of alkenes with readily available Ag(O2CCF2SO2F). | |
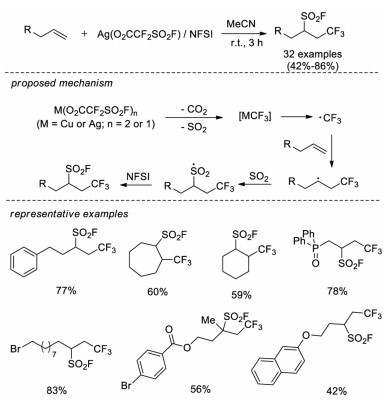{kind=link}
In 2018, the same group achieved radical fluoroalkylfluorosulfonylation reaction of unactivated alkenes [63]. Silver fluoroalkyl complexes (AgRF) were in situ generated from (fluoroalkyl)trimethylsilane and silver fluoride, which could deliver the fluoroalkyl radical for addition to alkenes. Subsequently, the combination of DABSO and NFSI provided the fluorosulfonyl source to trap the alkyl radical (Scheme 44).

|
Download:
|
| Scheme 44. Intermolecular oxidative radical fluoroalkylfluorosulfonylation of unactivated alkenes. | |
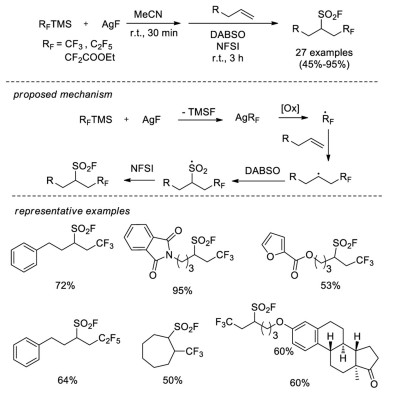{kind=link}
In the same year, Liu, Chen and co-workers developed another method to obtain fluoroalkyl radical for fluoroalkylfluorosulfonylation reaction of unactivated alkenes. The fluoroalkyl radical was generated from fluoroalkyl bromides (RFBr) under the reduction of zinc [64]. Both alkene and alkyne substrates are tolerated in this reaction, and a series of fluoroalkyl bromides are examined (such as BrCF2CO2Et, BrCF3, BrCF2CON(Et)2, Br(CF2)5CF3). It was also proposed that sulfur dioxide anion radical might be formed through reduction of DABSO with Zn, followed by electrophilic fluorination to afford the desired fluorosulfonyl products (Scheme 45).

|
Download:
|
| Scheme 45. Intermolecular reductive radical fluoroalkylsulfination of unactivated alkenes or alkynes. | |
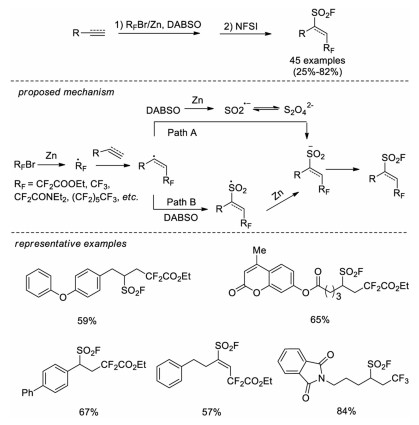{kind=link}
5. Synthesis of alkynyl sulfonyl fluorides (C(sp)-SO2F)
In 2020, Moses and co-workers developed a one-pot, stepwise approach for the synthesis of alkynyl sulfonyl fluorides from alkynes and the "SO2F" sources (FSO2-O-SO2F or SO2 + NFSI) [65]. A variety of 2-substituted-alkynyl-1-sulfonyl fluorides (SASF) were obtained in good yields (22 examples, 54%–88% yield). Combining the versatility of SuFEx click chemistry with classic C−C π-bond click chemistry, SASF were demonstrated to be a new class of connective hubs for diversity oriented clicking (DOC), and almost 300 novel compounds were rapidly prepared, including aromatic heterocyclic sulfonyl fluorides, bicyclic Diels-Alder adducts and sulfonates (Scheme 46). The potential of this DOC approach was further showcased through the identification of several lead hit compounds from an antibacterial screening against MRSA (USA300).

|
Download:
|
| Scheme 46. Synthesis of 2-substituted-alkynyl-1-sulfonyl fluorides. | |
{kind=link}
6. Conclusion
Sulfonyl fluorides, owing to their unique reactivity-stability balance, have found broad applications across an array of fields. In particular, since sulfur(VI) fluoride exchange (SuFEx) click chemistry was recently introduced by Sharpless and co-workers, there is an increasing demand for obtaining sulfonyl fluorides efficiently and practically. Herein we reviewed recent advances in the development of synthetic methods of sulfonyl fluorides, including aromatic, aliphatic, alkenyl, and alkynyl ones. Despite considerable efforts and substantial achievements, some notable issues still remain in this field:
(i) The source of SO2F group is limited. Although SO2F2 gas has served as a powerful and versatile SuFEx hub, it encounters several challenges as a reagent for use in research laboratories. ESF, another powerful connective hub, is fundamentally limited to the synthesis of sulfonyl fluorides with only C2 linkers. Moreover, despite the combination of a sulfur dioxide surrogate and a fluorine source provides alternative sources for assembling the SO2F groups, it suffers from low atom economy. Consequently, it is still desiredable to discover practical reagents for delivering SO2F moiety with high efficiency [66].
In comparison with aromatic sulfonyl fluorides, synthetic approaches for aliphatic, alkenyl, and alkynyl sulfonyl fluorides are relatively less explored. Expanding the scope will undoubtedly enable the application of these sulfonyl fluorides.
The asymmetric synthesis of chiral sulfonyl fluorides is less reported and still in its infancy. Given that chiral molecules possessing opposite configuration usually have distinct biological effects, future attention will be paid to this direction by designing suitable asymmetric catalytic systems.
(iv) While some progress has been made, utilization of novel synthetic strategies, such as direct C-H functionalizations, late-stage transformations and radical reactions, to regioselectively install a SO2F motif in drug-like molecules remains limited.
There is no doubt that the synthetic methods and application of sulfonyl fluorides will continue to grow in the near future. We hope this short review will arouse intensive research interest in sulfonyl fluoride chemistry and help chemists to apply the developed methodologies in related fields.
Declaration of competing interestThe authors declared that they do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21502240, 81972824), Guangdong Basic and Applied Basic Research Foundation (Nos. 2020A1515010684, 2020A1515011513), and Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (No. 2019B030301005).
| [1] |
(a) A.S. Barrow, C.J. Smedley, Q. Zheng, et al., Chem. Soc. Rev. 48 (2019) 4731-4758; (b) P.K. Chinthakindi, P.I. Arvidsson, Eur. J. Org. Chem. 2018 (2018) 3648-3666; (c) T. Abdul Fattah, A. Saeed, F. Albericio, J. Fluorine Chem. 213 (2018) 87-112. |
| [2] |
W. Steinkopf, J. Prakt. Chem. 117 (1927) 1-82. DOI:10.1002/prac.19271170101 |
| [3] |
J. Dong, L. Krasnova, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 53 (2014) 9430-9448. DOI:10.1002/anie.201309399 |
| [4] |
(a) P. Mukherjee, C.P. Woroch, L. Cleary, et al., Org. Lett. 20 (2018) 3943-3947; (b) H. Mukherjee, J. Debreczeni, J. Breed, et al., Org. Biomol. Chem. 15 (2017) 9685-9695; (c) S. Berg, M. Bergh, S. Hellberg, et al., J. Med. Chem. 55 (2012) 9107-9119. |
| [5] |
(a) M.K. Nielsen, C.R. Ugaz, W. Li, A.G. Doyle, J. Am. Chem. Soc. 137 (2015) 9571-9574; (b) J. Yin, D.S. Zarkowsky, D.W. Thomas, M.M. Zhao, M.A. Huffman, Org. Lett. 6 (2004) 1465-1468; (c) M.K. Nielsen, D.T. Ahneman, O. Riera, A.G. Doyle, J. Am. Chem. Soc. 140 (2018) 5004-5008. |
| [6] |
(a) J.A.H. Inkster, K. Liu, S. Ait-Mohand, et al., Chem. Eur. J. 18 (2012) 11079-11087; (b) L. Matesic, N.A. Wyatt, B.H. Fraser, et al., J. Org. Chem. 78 (2013) 11262-11270. |
| [7] |
G. Meng, T. Guo, T. Ma, et al., Nature 574 (2019) 86-89. DOI:10.1038/s41586-019-1589-1 |
| [8] |
(a) J. Dong, K.B. Sharpless, L. Kwisnek, J.S. Oakdale, V.V. Fokin, Angew. Chem. Int. Ed. 53 (2014) 9466-9470; (b) H. Wang, F. Zhou, G. Ren, et al., Angew. Chem. Int. Ed. 56 (2017) 11203-11208; (c) B. Gao, L. Zhang, Q. Zheng, et al., Nat. Chem. 9 (2017) 1083-1088; (d) C. Yang, J.P. Flynn, J. Niu, Angew. Chem. Int. Ed. 57 (2018) 16194-16199. |
| [9] |
(a) A. Narayanan, L.H. Jones, Chem. Sci. 6 (2015) 2650-2659; (b) E.C. Hett, H. Xu, K.F. Geoghegan, et al., ACS Chem. Biol. 10 (2015) 1094-1098; (c) O. Fadeyi, M.D. Parikh, M.Z. Chen, et al., ChemBioChem 17 (2016) 1925-1930; (d) L.H. Jones, ACS Med. Chem. Lett. 9 (2018) 584-586; (e) A.J. Brouwer, A. Jonker, P. Werkhoven, et al., J. Med. Chem. 55 (2012) 10995-11003; (f) C. Dubiella, H. Cui, M. Gersch, et al., Angew. Chem. Int. Ed. 53 (2014) 11969-11973; (g) N. Herrero Alvarez, H. van de Langemheen, A.J. Brouwer, R.M.J. Liskamp, Biorg. Med. Chem. 25 (2017) 5055-5063. |
| [10] |
(a) W. Davies, J.H. Dick, J. Chem. Soc. (1931) 2104-2109; (b) W. Davies, J.H. Dick, J. Chem. Soc. (1932) 483-486. |
| [11] |
T.A. Bianchi, L.A. Cate, J. Org. Chem. 42 (1977) 2031-2032. DOI:10.1021/jo00431a054 |
| [12] |
(a) G.J. Shafer, F. Forohar, D.D. DesMarteau, J. Fluorine Chem. 101 (2000) 27-29; (b) D.W. Kim, H.J. Jeong, S.T. Lim, et al., J. Org. Chem. 73 (2008) 957-962. |
| [13] |
(a) S.W. Wright, K.N. Hallstrom, J. Org. Chem. 71 (2006) 1080-1084; (b) G. Laudadio, A.A. Bartolomeu, L. Verwijlen, et al., J. Am. Chem. Soc. 141 (2019) 11832-11836; (c) Y. Cao, B. Adriaenssens, Ade A. Bartolomeu, et al., J. Flow Chem. 10 (2020) 191-197. |
| [14] |
(a) M. Kirihara, S. Naito, Y. Ishizuka, H. Hanai, T. Noguchi, Tetrahedron Lett. 52 (2011) 3086-3089; (b) M. Kirihara, S. Naito, Y. Nishimura, et al., Tetrahedron 70 (2014) 2464-2471. |
| [15] |
(a) M. Kulka, J. Am. Chem. Soc. 72 (1950) 1215-1218; (b) J.G. Kim, D.O. Jang, Synlett 2010 (2010) 3049-3052; (c) Y. Jiang, N.S. Alharbi, B. Sun, H.L. Qin, RSC Adv. 9 (2019) 13863-13867. |
| [16] |
(a) A.J. Brouwer, T. Ceylan, Tvd. Linden, R.M.J. Liskamp, Tetrahedron Lett. 50 (2009) 3391-3393; (b) A.J. Brouwer, T. Ceylan, A.M. Jonker, T. van der Linden, R.M. Liskamp, Bioorg. Med. Chem. 19 (2011) 2397-2406. |
| [17] |
(a) F. Toulgoat, B.R. Langlois, M. Médebielle, J.Y. Sanchez, J. Org. Chem. 72 (2007) 9046-9052; (b) L. Tang, Y. Yang, L. Wen, X. Yang, Z. Wang, Green Chem. 18 (2016) 1224-1228. |
| [18] |
M. Pérez-Palau, J. Cornella, Eur. J. Org. Chem. 2020 (2020) 2497-2500. DOI:10.1002/ejoc.202000022 |
| [19] |
A. Gomez-Palomino, J. Cornella, Angew. Chem. Int. Ed. 58 (2019) 18235-18239. DOI:10.1002/anie.201910895 |
| [20] |
A.T. Davies, J.M. Curto, S.W. Bagley, M.C. Willis, Chem. Sci. 8 (2017) 1233-1237. DOI:10.1039/C6SC03924C |
| [21] |
A.L. Tribby, I. Rodriguez, S. Shariffudin, N.D. Ball, J. Org. Chem. 82 (2017) 2294-2299. DOI:10.1021/acs.joc.7b00051 |
| [22] |
J. Kwon, B.M. Kim, Org. Lett. 21 (2019) 428-433. DOI:10.1021/acs.orglett.8b03610 |
| [23] |
C. Lee, N.D. Ball, G.M. Sammis, Chem. Commun. 55 (2019) 14753-14756. DOI:10.1039/C9CC08487H |
| [24] |
Y. Liu, D. Yu, Y. Guo, et al., Org. Lett. 22 (2020) 2281-2286. DOI:10.1021/acs.orglett.0c00484 |
| [25] |
T. Zhong, M.K. Pang, Z.D. Chen, et al., Org. Lett. 22 (2020) 3072-3078. DOI:10.1021/acs.orglett.0c00823 |
| [26] |
(a) Q. Lin, Z. Ma, C. Zheng, et al., Chin. J. Chem. 38 (2020) 1107-1110; (b) S. Liu, Y. Huang, X.H. Xu, F.L. Qing, J. Fluorine Chem. 240 (2020) 109653. |
| [27] |
(a) R.M. Hedrick, US Patent 2653973, 1953. (b) J.J. Krutak, R.D. Burpitt, W.H. Moore, J.A. Hyatt, J. Org. Chem. 44 (1979) 3847-3858. |
| [28] |
Y.P. Meng, S.M. Wang, W.Y. Fang, et al., Synthesis 52 (2019) 673-687. |
| [29] |
Q. Chen, P. Mayer, H. Mayr, Angew. Chem. Int. Ed. 55 (2016) 12664-12667. DOI:10.1002/anie.201601875 |
| [30] |
Q. Zheng, J. Dong, K.B. Sharpless, J. Org. Chem. 81 (2016) 11360-11362. DOI:10.1021/acs.joc.6b01423 |
| [31] |
H.L. Qin, Q. Zheng, G.A. Bare, P. Wu, K.B. Sharpless, Angew. Chem. Int. Ed. 55 (2016) 14155-14158. DOI:10.1002/anie.201608807 |
| [32] |
P.K. Chinthakindi, K.B. Govender, A.S. Kumar, et al., Org. Lett. 19 (2017) 480-483. DOI:10.1021/acs.orglett.6b03634 |
| [33] |
G.F. Zha, G.A.L. Bare, J. Leng, et al., Adv. Synth. Catal. 359 (2017) 3237-3242. DOI:10.1002/adsc.201700688 |
| [34] |
G.F. Zha, Q. Zheng, J. Leng, et al., Angew. Chem. Int. Ed. 56 (2017) 4849-4852. DOI:10.1002/anie.201701162 |
| [35] |
S.M. Wang, C. Li, J. Leng, S.N.A. Bukhari, H.L. Qin, Org. Chem. Front. 5 (2018) 1411-1415. DOI:10.1039/C7QO01128H |
| [36] |
(a) S.M. Wang, B. Moku, J. Leng, H.L. Qin, Eur. J. Org. Chem. 2018 (2018) 4407-4410; (b) C. Li, S.M. Wang, H.L. Qin, Org. Lett. 20 (2018) 4699-4703. |
| [37] |
G. Ncube, M.P. Huestis, Organometallics 38 (2019) 76-80. DOI:10.1021/acs.organomet.8b00327 |
| [38] |
X.Y. Chen, Y. Wu, J. Zhou, P. Wang, J.Q. Yu, Org. Lett. 21 (2019) 1426-1429. DOI:10.1021/acs.orglett.9b00165 |
| [39] |
(a) P.K. Mykhailiuk, Chem. Eur. J. 20 (2014) 4942-4947; (b) E.Y. Slobodyanyuk, O.S. Artamonov, O.V. Shishkin, P.K. Mykhailiuk, Eur. J. Org. Chem. 2014 (2014) 2487-2495. |
| [40] |
H. Jangra, Q. Chen, E. Fuks, et al., J. Am. Chem. Soc. 140 (2018) 16758-16772. DOI:10.1021/jacs.8b09995 |
| [41] |
J. Leng, H.L. Qin, Chem. Commun. 54 (2018) 4477-4480. DOI:10.1039/C8CC00986D |
| [42] |
(a) C.J. Smedley, M.C. Giel, A. Molino, et al., Chem. Commun. 54 (2018) 6020-6023; (b) J. Thomas, V.V. Fokin, Org. Lett. 20 (2018) 3749-3752. |
| [43] |
J. Leng, N.S. Alharbi, H.L. Qin, Eur. J. Org. Chem. 2019 (2019) 6101-6105. DOI:10.1002/ejoc.201901106 |
| [44] |
J. Leng, W. Tang, W.Y. Fang, C. Zhao, H.L. Qin, Org. Lett. 22 (2020) 4316-4321. DOI:10.1021/acs.orglett.0c01360 |
| [45] |
T.S.B. Lou, S.W. Bagley, M.C. Willis, Angew. Chem. Int. Ed. 58 (2019) 18859-18863. DOI:10.1002/anie.201910871 |
| [46] |
X. Nie, T. Xu, J. Song, et al., Angew. Chem. Int. Ed. 60 (2020) 3956-3960. |
| [47] |
A. Shavnya, S.B. Coffey, K.D. Hesp, S.C. Ross, A.S. Tsai, Org. Lett. 18 (2016) 5848-5851. DOI:10.1021/acs.orglett.6b02894 |
| [48] |
A. Shavnya, K.D. Hesp, A.S. Tsai, Adv. Synth. Catal. 360 (2018) 1768-1774. DOI:10.1002/adsc.201800071 |
| [49] |
X.R. Li, H.J. Chen, W. Wang, et al., J. Organomet. Chem. 899 (2019) 120912. DOI:10.1016/j.jorganchem.2019.120912 |
| [50] |
B. Moku, W.Y. Fang, J. Leng, E.A.B. Kantchev, H.L. Qin, ACS Catal. 9 (2019) 10477-10488. DOI:10.1021/acscatal.9b03640 |
| [51] |
B. Moku, W.Y. Fang, J. Leng, et al., iScience 21 (2019) 695-705. DOI:10.1016/j.isci.2019.10.051 |
| [52] |
J. Chen, B.Q. Huang, Z.Q. Wang, X.J. Zhang, M. Yan, Org. Lett. 21 (2019) 9742-9746. DOI:10.1021/acs.orglett.9b03911 |
| [53] |
J. Chen, D.Y. Zhu, X.Q. Zhang, M. Yan, J. Org. Chem. 86 (2021) 3041-3048. DOI:10.1021/acs.joc.0c02511 |
| [54] |
H.R. Chen, Z.Y. Hu, H.L. Qin, H. Tang, Org. Chem. Front. 8 (2021) 1185-1189. DOI:10.1039/D0QO01430C |
| [55] |
H.U. Daeniker, J. Druey, Helv. Chim. Acta 45 (1962) 1972-1981. DOI:10.1002/hlca.19620450633 |
| [56] |
J. Chanet-Ray, R. Vessiere, A. Zeroual, Heterocycles 26 (1987) 101-108. DOI:10.3987/R-1987-01-0101 |
| [57] |
Y.A. Skalenko, T.V. Druzhenko, A.V. Denisenko, et al., J. Org. Chem. 83 (2018) 6275-6289. DOI:10.1021/acs.joc.8b00077 |
| [58] |
V.L. Mykhalchuk, V.S. Yarmolchuk, R.O. Doroschuk, A.A. Tolmachev, O.O. Grygorenko, Eur. J. Org. Chem. 2018 (2018) 2870-2876. DOI:10.1002/ejoc.201800521 |
| [59] |
J. Liu, S.M. Wang, H.L. Qin, Org. Biomol. Chem. 18 (2020) 4019-4023. DOI:10.1039/D0OB00814A |
| [60] |
R. Xu, T. Xu, M. Yang, T. Cao, S. Liao, Nat. Commun. 10 (2019) 3752. DOI:10.1038/s41467-019-11805-6 |
| [61] |
X. Zhang, W.Y. Fang, R. Lekkala, W. Tang, H.L. Qin, Adv. Synth. Catal. 362 (2020) 3358-3363. DOI:10.1002/adsc.202000515 |
| [62] |
Y. Liu, H. Wu, Y. Guo, et al., Angew. Chem. Int. Ed. 56 (2017) 15432-15435. DOI:10.1002/anie.201709663 |
| [63] |
Q. Lin, Y. Liu, Z. Xiao, et al., Org. Chem. Front. 6 (2019) 447-450. DOI:10.1039/C8QO01192C |
| [64] |
Y. Liu, Q. Lin, Z. Xiao, et al., Chem. Eur. J. 25 (2019) 1824-1828. DOI:10.1002/chem.201805526 |
| [65] |
C.J. Smedley, G. Li, A.S. Barrow, et al., Angew. Chem. Int. Ed. 59 (2020) 12460-12469. DOI:10.1002/anie.202003219 |
| [66] |
(a) X. Zhang, B. Moku, J. Leng, K.P. Rakesh, H.L. Qin, Eur. J. Org. Chem. 2019 (2019) 1763-1769; (b) W.Y. Fang, S.M. Wang, Z.W. Zhang, H.L. Qin, Org. Lett. 22 (2020) 8904-8909; (c) Z.W. Zhang, S.M. Wang, W.Y. Fang, R. Lekkala, H.L. Qin, J. Org. Chem. 85 (2020) 13721-13734. |