 2021, Vol. 32
2021, Vol. 32 


b Faculty of Environment and Life, Beijing University of Technology, Beijing 100024, China;
c College of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang 473061, China
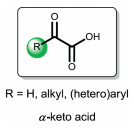
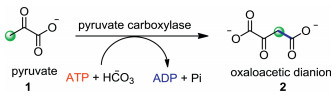
α-Keto acids are a class of organic compounds containing a keto function adjacent to a carboxylic acid group (Fig. 1). α-Keto acids are wildly occurred in the energy-supplying biochemical processes [1, 2]. For example, pyruvate 1 is an important metabolite in the Krebs cycle, also called tricarboxylic acid cycle, which triggers a number of enzyme-catalyzed reactions to give adenosine triphosphate (ATP). Pyruvate 1 can also undergo condensation reaction with carbonic acid utilizing pyruvate carboxylase as the catalyst, affording dianion 2 which is another central metabolite in the Krebs cycle (Scheme 1). Considering the importance of α-keto acids in biochemical processes, the discovery of new chemical properties of α-keto acids is of great significance. Since Gooßen et al. reported the first example of decarboxylative coupling of aryl bromides with α-keto acids, the use of α-keto acids as green acyl surrogates has been extensively investigated since this type of reactions excluded nontoxic CO2 as the by-product [3]. Recently, some nice reviews have highlighted the advances in metal- or photo-catalyzed decarboxylative couplings of α-keto acids [4, 5].

|
Download:
|
| Fig. 1. The structure of α-keto acids. | |
{kind=link}

|
Download:
|
| Scheme 1. Biochemical transformation of pyruvate. | |
{kind=link}
Organic electrosynthesis employs electrons as the clean redox reagents, thus avoiding the use of harmful and dangerous chemical redox reagents [6-13]. Apart from the advantages of reducing byproduct generation and improving the cost-efficiency of synthetic processes, organic electrochemistry has the ability to access reactive intermediates under mild conditions, which are challenging for conventional approaches. Recently, organic electrosynthesis has experienced a renaissance after being overlooked by synthetic chemists for many years [14-25]. Inspired by the merits of organic electrosynthesis, many synthetic applications of α-keto acids under electrochemical conditions have been described in recent years. However, there is no systematic review dealing with this topic. Herein, we present the recent breakthroughs achieved in the electrochemical transformations of α-keto acids. This review can be classified into three categories: electrochemical decarboxylative acylation reactions, electrochemical decarboxylative cyclization reactions, and electrochemical reductive amination reactions. In addition to the description of advantages of these methodologies, we also want to point out the limitations to the readers.
2. Electrochemical decarboxylative acylation reactionsMinisci acylation reaction provides a facile route to obtain valuable carbonyl-substituted N-heterocycles, which is challenging for Friedel-Crafts acylation reactions [26-29]. Typically, Minisci acylation reactions employed Fe or Ag salts as the catalysts and (NH4)2S2O8 as the oxidant [30, 31]. In 2017, we reported an electrochemical decarboxylative Minisci acylation reaction under metal- and chemical oxidant-free conditions with α-keto acids as acyl surrogates (Scheme 2) [32]. This reaction was carried out in an undivided cell with graphite as the electrodes, LiClO4/CH3CN as the electrolytic solution, readily available NH4I as the catalyst, and hexafluoroisopropanol (HFIP) as the additive under controlled-current electrolytic (CCE) conditions. A diverse of N-heterocycles such as quinoxaline (5a, 5g), pyrazine (5b, 5c), pyridazine (5d), pyridine (5e), and quinoline (5f) were all suitable substrates under the electrolytic conditions. Besides, aliphatic and aromatic α-keto acids were all well-tolerated under the standard conditions. However, this methodology still has some limitations: 1) most of the N-heterocycles showed moderate efficiencies; 2) electron-withdrawing α-keto acid did not work under the optimal conditions (5h).

|
Download:
|
| Scheme 2. NH4I-catalyzed electrochemical Minisci acylation reaction. | |
{kind=link}
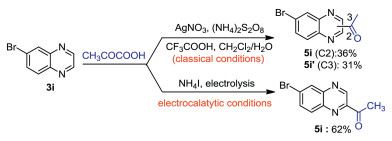
One of the advantages of this electrochemical protocol is the controllable formation of acyl radical under a relative low concentration. However, for chemical oxidant-mediated Minisci acylation reactions, acyl radical formation was finished in a short time. The high concentration of acyl radicals in the reaction mixture would lead to radical decomposition and poor selectivities. To demonstrate the selectivity advantage of this electrochemical protocol over traditional versions, the Minisci acylation of 3i was carried out under different conditions. As shown in Scheme 3, when the reaction was carried out under Ag/(NH4)2S2O8-mediated conditions, the acylation occurred at C2 and C3 positions; however, the acylation only occurred at C2 position under electrocatalytic conditions.

|
Download:
|
| Scheme 3. Comparison of selectivities between conventional and electrochemical conditions. | |
{kind=link}
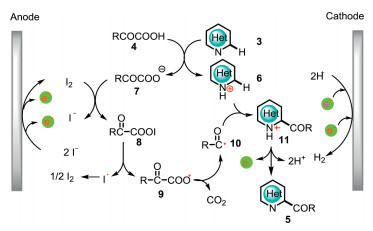
The plausible mechanism for the electrochemical Minisci acylation reaction was shown in Scheme 4. Initially, the reaction between N-heterocycle 3 and α-keto acid 4 gives protonated N-heterocycle 6 and carboxylate anion 7. Then, carboxylate anion 7 reacts with anodically generated iodine to afford acyl hypoiodite 8, which undergoes a homolytic cleavage to produce radical 9 and iodine radical. Aroyloxy radical 9 subsequently excludes CO2 to give acyl radical 10, which has a radical addition to 6 affording 11. Finally, intermediate 11 undergoes a further oxidation and subsequently releases protons to give acylated product 5. The controllable generation of acyl hypoiodite 8 as a mask of acyl radical is the key to the mono-selectivity of this electrochemical Minisci acylation reaction. The employment of HFIP as the additive not only assists the cathodic hydrogen evolution, but also stabilizes the generated radical species.

|
Download:
|
| Scheme 4. Mechanistic proposal for the formation of 5. | |
{kind=link}
To overcome the substrate scope limitations of the above-mentioned methodology (Scheme 2), we want to develop a new catalytic system to facilitate the acyl radical formations from electron-deficient α-keto acids. In 2019, we reported the first example of Ni-catalyzed electrochemical Minisci acylation reaction with α-keto acids as acyl surrogates (Scheme 5) [33]. This reaction was carried out in an undivided cell equipped with Pt anode and graphite cathode with n-Bu4NBF4/DCM/CH3CN as the electrolytic solution under CCE conditions. Electron-deficient α-keto acids and acridine were well-tolerated under this newly developed catalytic system. However, this methodology has its own limitations: 1) phenanthridine gave nonaromatic 14d as the byproduct under the optimal conditions; 2) isoquinoline (14h) and benzothiazole (14i) did not work under the standard conditions.

|
Download:
|
| Scheme 5. Nickel-catalyzed electrochemical Minisci acylation reaction. | |
{kind=link}
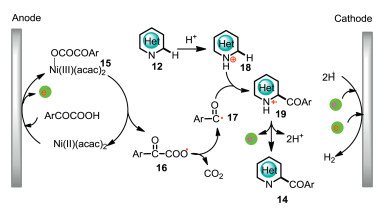
Control experiments showed that nickel catalyst is crucial for the decarboxylative formation of acyl radicals. Cyclic voltammetry (CV) experiments revealed that the oxidation potential of the complex of Ni(acac)2 and 2-oxo-2-phenylacetic acid has a negative shift of 0.19V compared with the oxidation potential of Ni(acac)2. Based on the control experiments and CV studies, a plausible mechanism was shown in Scheme 6. The complex of Ni(acac)2 and α-keto acids initially undergoes an anodic oxidation to give hypervalent nickel complex 15, which has an inner-sphere electron transfer to produce aroyloxy radical 16. Subsequent decarboxylation and radical addition afford radical cation 19, which has further anodic oxidation followed by protons releasing to give valuable acylated N-heterocycle 14. The hypervalent nickel complex 15 is a mask of acyl radical, which can afford acyl radical in a controllable concentration under electrolytic conditions.

|
Download:
|
| Scheme 6. Mechanistic proposal for the formation of 14. | |
{kind=link}
Enaminones are valuable structural motifs in natural products and pharmaceutically important compounds [34, 35]. The construction of enaminone in a green manner is of great significance. In 2019, with acyl hypoiodite 8 as the mask of acyl radical as described in Scheme 4, Chen and Xu developed an electrochemical acylation of vinyl azides 20 with α-keto acids 21 (Scheme 7) [36]. This electrochemical protocol provides an environmentally benign route to enaminones, while previous report for the acylation of vinyl azides with α-keto acids needed CuI as the catalyst [37]. This decarboxylative coupling reaction was carried out in an undivided cell equipped with graphite anode and Pt cathode with n-Bu4NI as the catalyst under CCE conditions. Under the optimal conditions, a diverse of electron-withdrawing vinyl azides were tolerated well to give enaminones in moderate yields (22a–22c). Heteroaryl substituted vinyl azides were also suitable substrates, affording the corresponding enaminones in moderate yields (22d–22f). Moreover, aliphatic α-keto acids underwent the acylation reaction smoothly with lower yields (22g–22i). However, the alkylated vinyl azide did not work under the standard conditions (22j).

|
Download:
|
| Scheme 7. NH4I-catalyzed electrochemical acylation reaction. | |
{kind=link}
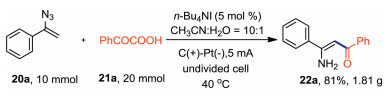
The scale-up experiment demonstrated the practical utility of this electrochemical acylation reaction. As shown in Scheme 8, when the acylation reaction of 20a and 21a was carried out on a 10mmol scale, the valuable acylated product 22a was isolated in 81% yield.

|
Download:
|
| Scheme 8. Scale-up experiment. | |
{kind=link}
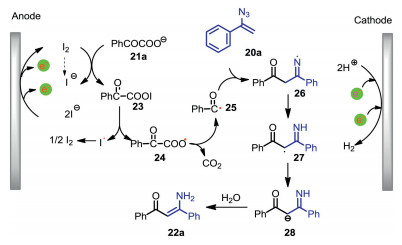
The plausible mechanism for the formation of 22a was shown in Scheme 9. First, α-keto carboxylate anion 21a reacts with anodically generated iodine to give acyl hypoiodite 23, which undergoes homolytic cleavage and following decarboxylation to afford acyl radical 25. Radical addition of 25 to vinyl azide 20a gives radical 26, which triggers an intramolecular hydrogen atom abstraction to produce carbon-centered radical 27. Finally, further reduction of radical 27 followed by protonation affords the desired enaminone 22a. Acyl hypoiodite 23 is a key intermediate for this electrochemical acylation reaction.

|
Download:
|
| Scheme 9. A proposed reaction pathway for the formation of 22a. | |
{kind=link}
Formamides are always employed as important intermediates for the construction of value-added products in organic synthesis [38, 39]. Besides, formamides are widely existed in many pharmaceutically relevant compounds [40, 41]. As glyoxylic acid is readily available and stable, the decarboxylative coupling of glyoxylic acid with amines would provide an appealing alternative for the construction of formamides, since the reaction generates CO2 as the sole byproduct. In 2018, Huang and co-workers realized an electrochemical N-formylation of diverse amines with the smallest α-keto acids, glyoxylic acid [42]. The electrochemicalN-formylation reaction was carried out in an undivided cell equipped with Pt electrodes with Cu(II) and Ni(II) as the catalysts under CCE conditions. As shown in Scheme 10, this protocol could give various formamides in moderate to excellent yields. First, acyclic and cyclic secondary amines could be transformed into the corresponding formamides efficiently (31a–31d). Second, primary amines including aniline and heterocyclic amines were all compatible under the optimal conditions (31e–31g). However, aliphatic amines gave lower yields (31d, 31h) owing to their low efficiencies of the corresponding condensation reactions with glyoxylic acid.

|
Download:
|
| Scheme 10. Electrochemical N-formylation of amines with glyoxylic acid. | |
{kind=link}
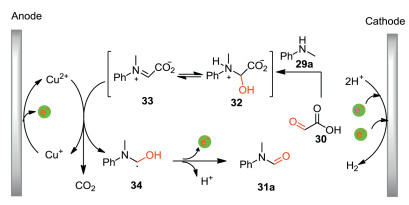
The plausible reaction pathway for the formation of formamide 31a was shown in Scheme 11. Initially, the condensation of N-methylaniline 29a with glyoxylic acid 30 gives intermediate 32, which is oxidized by Cu(II) followed by decarboxylation to produce radical 34. Meanwhile, Cu(I) is generated which can be re-oxidized to Cu(II) at the anode. Subsequently, radical 34 undergoes further single electron oxidation followed by proton releasing to give formamide 31a. The Cu(II) species acts as an active oxidant which can be supported by the observation of catalytic current of Cu(II) catalyst in the CV experiments.

|
Download:
|
| Scheme 11. A plausible reaction pathway for the formation of 31a. | |
{kind=link}
3. Electrochemical decarboxylative cyclization reactions
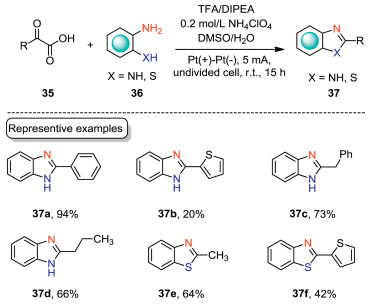
2-Substituted benzimidazoles are privileged building blocks for various natural products, and pharmaceutically important compounds [43, 44]. Consequently, much effort has been devoted to the construction of 2-substituted benzimidazoles. In 2016, Huang and co-workers reported an electrochemical decarboxylative cyclization of diamines with α-keto acids to construct 2-substituted benzimidazoles (Scheme 12) [45]. This cyclization reaction was carried out in an undivided cell equipped with Pt electrodes with NH4ClO4/DMSO/H2O as the electrolytic solution under CCE conditions. Aromatic α-keto acid underwent the decarboxylative cyclization smoothly to yield 2-substituted benzimidazole in excellent yield (37a). Aliphatic α-keto acids were also suitable substrates under the optimal conditions (37c, 37d). Moreover, benzothiazole could also be obtained by this electrochemical protocol (37e). However, heteroaromatic α-keto acids as the substrates only gave the corresponding products in low yields (37b, 37f).

|
Download:
|
| Scheme 12. Electrochemical decarboxylative cyclization of α-keto acids. | |
{kind=link}
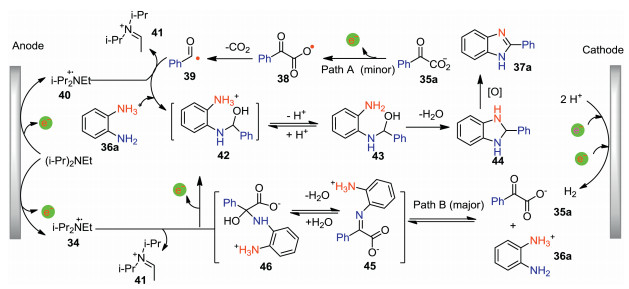
The plausible reaction pathway for the formation of benzimidazole 37a was shown in Scheme 13. This decarboxylative cyclization proceeded via two pathways (path A and path B). For path A, 2-oxo-2-phenylacetic acid 35a undergoes anodic oxidation and subsequent decarboxylation to give acyl radical 39, which reacts with protonated diamine 36a and then abstracts a hydrogen atom from anodically generated radical cation 40 to afford intermediate 42. For path B, the condensation of 35a and 36a gives hemiaminal 46, which undergoes decarboxylation and further H atom abstraction from anodically generated radical cation 40 to afford intermediate 42. Then, intermediate 42 loses one molecule of proton followed by intramolecular cyclization to give 44, which has further oxidation to produce benzimidazole 37a. Previous report showed that the decarboxylation of hemiaminal 46 has a lower activation barrier than 2-oxo-2-phenylacetic acid 35a [46]. Besides, iminocarboxylate 45 was detected during the electrolysis. Therefore, the authors believed that path B is preferable to path A.

|
Download:
|
| Scheme 13. A tandem reaction pathway for the formation of 37a. | |
{kind=link}
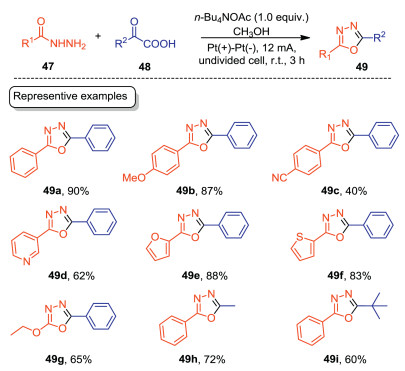
1, 3, 4-Oxadiazoles are widely existed in many pharmaceutically valuable agents and functional materials [47, 48]. Thus, the green construction of 1, 3, 4-oxadiazoles has attracted much attention in recent years. In 2020, Gao, Zhang, Lei and co-workers developed an electrochemical decarboxylative cyclization of acylhydrazines with α-keto acids to furnish 1, 3, 4-oxadiazoles with high efficiencies (Scheme 14) [49]. The reaction was carried out in an undivided cell equipped with Pt electrodes with n-Bu4NOAc/CH3OH as the electrolytic solution under CCE conditions. For acylhydrazines, both aromatic and heteroaromatic acylhydrazines underwent the electrochemical cyclization smoothly to give the corresponding 1, 3, 4-oxadiazoles in good to excellent yields (49a–49f). Aliphatic acylhydrazine was also suitable substrate, giving 1, 3, 4-oxadiazole in 65% yield (49g). For α-keto acids, both aromatic and aliphatic α-keto acids were tolerated well under the optimal conditions. Compared with the previous reports dealing with 1, 3, 4-oxadiazoles syntheses, this protocol employed electron as the clean redox reagent, thus providing an appealing alternative for the green synthesis of 1, 3, 4-oxadiazoles.

|
Download:
|
| Scheme 14. Electrochemical synthesis of 1, 3, 4-oxadiazoles. | |
{kind=link}
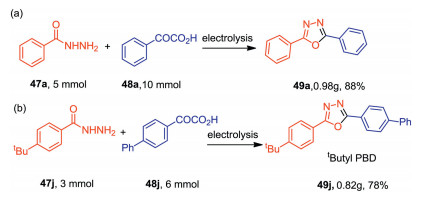
The practicability of this protocol was further demonstrated by the scale-up experiments. As shown in Scheme 15, the reactions can be carried out on a 5mmol scale with high efficiencies. Moreover, compound 49j which is important in the field of liquid scintillator neutrino detector can be obtained on a 0.82g scale.

|
Download:
|
| Scheme 15. Scale-up experiments. | |
{kind=link}
The plausible reaction pathway for the formation of 1, 3, 4-oxadiazole 49a was shown in Scheme 16. First, the condensation of 47a and 48a gives imine 50, which is deprotonated by cathodically generated MeO– affording carboxylate anion 51. Then, intermediate 51 has an anodic oxidation and subsequent decarboxylation to yield radical 53, which undergoes further intramolecular cyclization to give intermediate 54. Finally, intermediate 54 loses one electron and one molecule of proton to yield 1, 3, 4-oxadiazole 49a. The proposed radical pathway and the formation of imine 50 have been demonstrated by the radical trapping experiments and control experiments.

|
Download:
|
| Scheme 16. A plausible reaction pathway for the formation of 49a. | |
{kind=link}
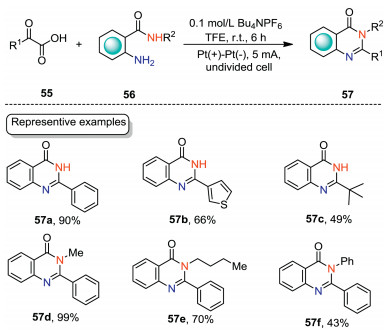
Quinazolinone skeleton represents one of the privileged structural motifs in natural alkaloids, and pharmaceutical reagents [50-52]. The construction of quinazolinones in an environmentally benign manner is one of the pursuits of organic chemists. In 2021, Wei and co-workers developed an electrochemical decarboxylative cyclization of 2-aminobenzamides with α-keto acids for the synthesis of quinazolin-4(3H)-ones under mild conditions (Scheme 17) [53]. The electrochemical cyclization was carried out in an undivided cell equipped with Pt electrodes with n-Bu4NPF6 in TFE as the electrolytic solution under CCE conditions. When R2 = H, 2-oxo-2-phenylacetaic acid, 2-oxo-2-(thiophen-3-yl)acetic acid, and aliphatic α-keto acid were all suitable substrates, affording the corresponding quinazolin-4(3H)-ones with up to 90% yield (57a–57c). When 2-amino-N-substituted benzamides were employed as the substrates, the reactions could also underwent the cyclization reaction smoothly (57d–57f).

|
Download:
|
| Scheme 17. Electrochemical synthesis of quinazolin-4(3H)-ones. | |
{kind=link}
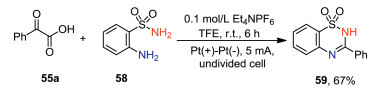
In addition to 2-aminobenzamide, 2-aminobenzensulfonamide 58 could also undergo decarboxylative cyclization with α-keto acid to give the desired product 59 in 67% yield (Scheme 18).

|
Download:
|
| Scheme 18. Electrochemical cyclization of 2-aminobenzensulfonamide with α-keto acid. | |
{kind=link}
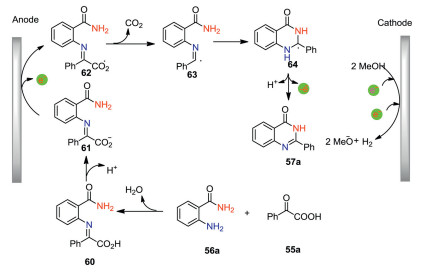
The plausible mechanism for the formation of 57a was shown in Scheme 19. The condensation of 55a with 56a gives imine 60, which releases one molecule of proton to yield carboxylate anion 61. The anodic oxidation of 61 followed by decarboxylation leads to the formation of radical 63, which undergoes an intramolecular cyclization to give carbon-centered radical 64. Finally, further anodic oxidation of radical 64 and subsequent deprotonation affords quinazolin-4(3H)-one. Compared with previous reports on quinazolin-4(3H)-ones syntheses, this electrochemical protocol features metal-, base- and external oxidant-free conditions. However, the use of toxic TFE as the solvent is the drawback of this protocol. Further investigations to avoid the use of toxic solvent would surely improve the synthetic potential of this methodology.

|
Download:
|
| Scheme 19. A mechanistic proposal for the formation of 57a. | |
{kind=link}
4. Electrochemical reductive amination reactions
Amino acids play central roles as intermediates in metabolism and as building blocks in nature [54, 55]. Considering the importance of amino acids, significant progress has been made on amino acids production. Among many synthetic protocols to produce amino acids, the most conventional approach for the chemical synthesis of amino acids is the Strecker reaction [56], which assembles amino acids from ammonia, cyanide, and aldehydes. However, the use of hazardous cyanide is the drawback of this reaction. To overcome this limitation, reductive amination of α-keto acids without consumption of hazardous reagents represents an appealing alternative to amino acid synthesis. Pioneered by Jeffery and co-workers [57, 58], the electrochemical reductive amination of α-keto acids in aqueous ammonia has been demonstrated to be a green approach for the synthesis of amino acids. As shown in Scheme 20, the electrolysis was carried out in a divided cell equipped with Pt electrodes in aqueous NH3/NH4Cl solution under constant potential conditions. 2-Oxo-2-phenylacetaic acid could give phenylglycine 67a in 83% yield, while pyruvic acid gave alanine 67b in lower yield. It is noteworthy that monoammonium glutamate 67c could also be obtained in 64% yield under the optimal conditions. However, phenylalanine 67d could not be produced by this method.

|
Download:
|
| Scheme 20. Electrochemical synthesis of α-amino acids. | |
{kind=link}
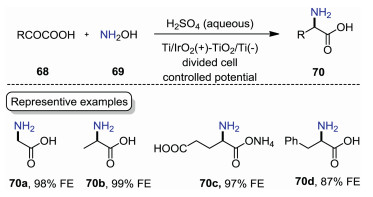
Even though the above-mentioned method could produce amino acids efficiently, the use of precious Pt electrodes limits its wide application. In 2019, Yamauchi and co-workers reported an electrochemical reductive amination of α-keto acids with NH2OH on titanium dioxide [59]. As shown in Scheme 21, the reaction was carried out in a divided cell equipped with titanium dioxide electrode in aqueous H2SO4 under constant potential conditions. Glycine 70a, alanine 70b, and monoammonium glutamate 70c were obtained with up to 99% Faraday efficiency (FE). Phenylalanine 70d which cannot be obtained in previous report was synthesized with a 87% FE. This protocol features high FE values, which would reduce the cost-efficiency of synthetic process for amino acids production.

|
Download:
|
| Scheme 21. Electrochemical reductive amination of α-keto acids on titanium dioxide. | |
{kind=link}
In conclusion, this review summarized the recent advances achieved in electrochemical transformations of α-keto acids. The introduction of electrochemistry further enriches the synthetic utilities of α-keto acids. α-Keto acids can be employed as green acyl surrogates in electrochemical acylation and cyclization reactions to afford value-added chemicals. Besides, electrochemical reductive amination of α-keto acids has been demonstrated to be an appealing alternative to produce amino acids, which are key building blocks in nature. Even though substantial progress has been made on electrochemical transformations of α-keto acids, some challenges still remain: 1) Further improvement of the efficiencies of heteroaromatic and aliphatic α-keto acids under electrochemical conditions would expand the synthetic applications of α-keto acids; 2) The electrosynthesis of amino acids via reductive aminations of α-keto acids in an undivided cell under CCE conditions would be another challenge in this avenue.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (No. 21702113), the Thousand Talents Plan of Central Plains, and Beijing Municipal Education Committee Project (No. KM202110005006).
| [1] |
J. Kay, P.D.J. Weitzman, Biochemical Society Symposia, Vol. 54, The Biochemical Society, London, 1987.
|
| [2] |
D. Voet, J.G., Voet, Biochemistry. 3rd ed.. New York: John Wiley & Sons, 2004.
|
| [3] |
L.J. Gooßen, F. Rudolphi, C. Oppel, N. Rodrguez, Angew. Chem. Int. Ed. 47 (2008) 3043-3045. DOI:10.1002/anie.200705127 |
| [4] |
Y. Wei, P. Hu, M. Zhang, W. Su, Chem. Rev. 117 (2017) 8864-8907. DOI:10.1021/acs.chemrev.6b00516 |
| [5] |
F. Penteado, E.F. Lopes, D. Alves, et al., Chem. Rev. 119 (2019) 7113-7278. DOI:10.1021/acs.chemrev.8b00782 |
| [6] |
S. Tang, Y.C. Liu, A.W. Lei, Chem 4 (2018) 27-45. DOI:10.1016/j.chempr.2017.10.001 |
| [7] |
Y.Y. Jiang, K. Xu, C.C. Zeng, Chem. Rev. 118 (2018) 4485-4540. DOI:10.1021/acs.chemrev.7b00271 |
| [8] |
L. Ackermann, Acc. Chem. Res. 53 (2020) 84-104. DOI:10.1021/acs.accounts.9b00510 |
| [9] |
P. Xiong, H.C. Xu, Acc. Chem. Res. 52 (2019) 3339-3350. DOI:10.1021/acs.accounts.9b00472 |
| [10] |
Z.H. Ye, F.Z. Zhang, Chin. J. Chem. 37 (2019) 513-528. DOI:10.1002/cjoc.201900049 |
| [11] |
K.J. Jiao, Y.K. Xing, Q.L. Yang, H. Qiu, T.S. Mei, Acc. Chem. Res. 53 (2020) 300-310. DOI:10.1021/acs.accounts.9b00603 |
| [12] |
J. Liu, L. Lu, D. Wood, S. Lin, ACS Cent. Sci. 6 (2020) 1317-1340. DOI:10.1021/acscentsci.0c00549 |
| [13] |
J. Zhong, Y. Yu, D. Zhang, K.Y. Ye, Chin. Chem. Lett. 32 (2021) 963-972. DOI:10.1016/j.cclet.2020.08.011 |
| [14] |
T.J. DeLano, S.E. Reisman, ACS Catal. 9 (2019) 6751-6754. DOI:10.1021/acscatal.9b01785 |
| [15] |
X. Liu, R. Liu, J. Qiu, X. Cheng, G. Li, Angew. Chem. Int. Ed. 59 (2020) 13962-13967. DOI:10.1002/anie.202005765 |
| [16] |
P. Xu, P.Y. Chen, H.C. Xu, Angew. Chem. Int. Ed. 59 (2020) 14275-14280. DOI:10.1002/anie.202005724 |
| [17] |
F. Wang, S.S. Stahl, Angew. Chem. Int. Ed. 58 (2019) 6385-6390. DOI:10.1002/anie.201813960 |
| [18] |
L. Capaldo, L.L. Quadri, D. Ravelli, Angew. Chem. Int. Ed. 58 (2019) 17508-17510. DOI:10.1002/anie.201910348 |
| [19] |
Z. Ye, Y. Wu, N. Chen, et al., Nat. Commun. 11 (2020) 3628. DOI:10.1038/s41467-020-17389-w |
| [20] |
L. Niu, C. Jiang, Y. Liang, et al., J. Am. Chem. Soc. 142 (2020) 17693-17702. DOI:10.1021/jacs.0c08437 |
| [21] |
S. Zhang, L.J. Li, J.J. Zhang, et al., Chem. Sci. 10 (2019) 3181-3185. DOI:10.1039/C9SC00100J |
| [22] |
H. Wang, J.X. Shi, J. Tan, et al., Org. Lett. 21 (2019) 9430-9433. DOI:10.1021/acs.orglett.9b03641 |
| [23] |
M.X. He, Z.Y. Mo, Z.Q. Wang, et al., Org. Lett. 22 (2020) 724-728. DOI:10.1021/acs.orglett.9b04549 |
| [24] |
J. Zhang, H. Wang, Y. Chen, et al., Chin. Chem. Lett. 31 (2020) 1576-1579. DOI:10.1016/j.cclet.2019.11.037 |
| [25] |
S. Zhang, L. Li, J. Shi, et al., Angew. Chem. Int. Ed. 60 (2021) 7275-7282. DOI:10.1002/anie.202015230 |
| [26] |
J. Tauber, D. Imbri, T. Opatz, Molecules 19 (2014) 16190-16222. DOI:10.3390/molecules191016190 |
| [27] |
W. Xiao, J. Wu, Chin. Chem. Lett. 31 (2020) 3083-3094. DOI:10.1016/j.cclet.2020.07.035 |
| [28] |
L. Liu, P. Jiang, Y. Liu, H. Du, J. Tan, Org. Chem. Front. 7 (2020) 2278-2283. DOI:10.1039/D0QO00507J |
| [29] |
S. Peng, Y.X. Song, J.Y. He, et al., Chin. Chem. Lett. 30 (2019) 2287-2290. DOI:10.1016/j.cclet.2019.08.002 |
| [30] |
L. Désaubry, J.J. Bourguignon, Tetrahedron Lett. 36 (1995) 7875-7876. DOI:10.1016/0040-4039(95)01647-Z |
| [31] |
F. Minisci, F. Recupero, A. Cecchetto, et al., Org. Process Res. Dev. 8 (2004) 163-168. DOI:10.1021/op034137w |
| [32] |
Q.Q. Wang, K. Xu, Y.Y. Jiang, et al., Org. Lett. 19 (2017) 5517-5520. DOI:10.1021/acs.orglett.7b02589 |
| [33] |
H. Ding, K. Xu, C.C. Zeng, J. Catal. 381 (2020) 38-43. DOI:10.1016/j.jcat.2019.10.030 |
| [34] |
B. Stanovnik, J. Svete, Chem. Rev. 104 (2004) 2433-2480. DOI:10.1021/cr020093y |
| [35] |
G. Negri, C. Kascheres, A.J. Kascheres, Heterocycl. Chem. 41 (2004) 413-491. DOI:10.1002/jhet.5570410318 |
| [36] |
X. Kong, Y. Liu, L. Lin, Q. Chen, B. Xu, Green Chem. 21 (2019) 3796-3801. DOI:10.1039/C9GC01098J |
| [37] |
Y. Ning, X.F. Zhao, Y.B. Wu, X. Bi, Org. Lett. 19 (2017) 6240-6243. DOI:10.1021/acs.orglett.7b03204 |
| [38] |
M. Hosseini-Sarvari, H. Sharghi, J. Org. Chem. 71 (2006) 652-6654. |
| [39] |
J.Y. Quek, T.P. Davis, A.B. Lowe, Chem. Soc. Rev. 42 (2013) 7326-7334. DOI:10.1039/c3cs60065c |
| [40] |
R. Hett, Q.K. Fang, Y. Gao, S.A. Wald, C.H. Senanayake, Org. Process Res. Dev. 2 (1998) 96-99. DOI:10.1021/op970116o |
| [41] |
G. Ma, M. Zancanella, Y. Oyola, et al., Org. Lett. 8 (2006) 4497-4500. DOI:10.1021/ol061651o |
| [42] |
D.Z. Lin, J.M. Huang, Org. Lett. 20 (2018) 2112-2115. DOI:10.1021/acs.orglett.8b00698 |
| [43] |
Y. Bansal, O. Silakari, Bioorg. Med. Chem. 20 (2012) 6208-6236. DOI:10.1016/j.bmc.2012.09.013 |
| [44] |
P. Singla, V. Luxami, K. Paul, RSC Adv. 4 (2014) 12422-12440. DOI:10.1039/c3ra46304d |
| [45] |
H.B. Wang, J.M. Huang, Adv. Synth. Catal. 358 (2016) 1975-1981. DOI:10.1002/adsc.201501167 |
| [46] |
F.B. Song, L.J. Gooßen, Adv. Synth. Catal. 353 (2011) 337-342. DOI:10.1002/adsc.201000798 |
| [47] |
L.H. Chan, R.H. Lee, C.F. Hsieh, H.C. Yeh, C.T. Chen, J. Am. Chem. Soc. 124 (2002) 6469-6479. DOI:10.1021/ja0255150 |
| [48] |
S. Valente, D. Trisciuoglio, T. De Luca, et al., J. Med. Chem. 57 (2014) 6259-6265. DOI:10.1021/jm500303u |
| [49] |
F. Lu, F. Gong, L. Li, et al., Eur. J. Org. 2020 (2020) 3257-3260. DOI:10.1002/ejoc.202000311 |
| [50] |
S.B. Mhaske, N.P. Argade, Tetrahedron 62 (2006) 9787-9826. DOI:10.1016/j.tet.2006.07.098 |
| [51] |
I. Khan, A. Ibrar, N. Abbas, A. Saeed, Eur. J. Med. Chem. 76 (2014) 193-244. DOI:10.1016/j.ejmech.2014.02.005 |
| [52] |
P.P. Kung, M.D. Casper, K.L. Cook, et al., J. Med. Chem. 42 (1999) 4705-4713. DOI:10.1021/jm9903500 |
| [53] |
Q. Tian, J. Zhang, L. Xu, Y. Wei, Mol. Catal. 500 (2021) 111345. DOI:10.1016/j.mcat.2020.111345 |
| [54] |
D.H. Baker, Amino Acids 37 (2009) 29-41. DOI:10.1007/s00726-008-0198-3 |
| [55] |
G. Wu, Amino Acids 37 (2009) 1-17. |
| [56] |
H. Groger, Chem. Rev. 103 (2003) 2795-2828. DOI:10.1021/cr020038p |
| [57] |
E.A. Jeffery, O. Johansen, A. Meisters, Aust. J. Chem. 31 (1978) 73-78. DOI:10.1071/CH9780073 |
| [58] |
E.A. Jeffery, O. Johansen, A. Meisters, Aust. J. Chem. 31 (1978) 79-84. DOI:10.1071/CH9780079 |
| [59] |
T. Fukushima, M. Yamauchi, Chem. Commun. 55 (2019) 14721-14724. DOI:10.1039/C9CC07208J |