 2021, Vol. 32
2021, Vol. 32 


A series of crises such as air pollution, energy shortages and global warming are driving by the ever-increasing consumption of fossil fuels, thus facilitating the rapid development of sustainable, environmentally friendly alternative energy sources [1-4]. Dealing with environmental pollution problems, searching clean, cost effectiveness and renewable energy resource is an urgent mission to achieve sustainable development [5-8]. Molecular hydrogen (H2) is considered to be one of the most promising fuels. With the highest gravitational energy density and its only combustion byproduct being non-polluting water, H2 is an excellent energy carrier and a potential candidate for future low-carbon energy systems [9-11]. Additionally, hydrogen element is the most abundant substance in the universe and largely stored in widespread water resource on the earth. Therefore, the renewable, pollution-free and large reserved of hydrogen energy may become the ultimate energy programmed in the future [12, 13].
Producing clean energy through steam methane reforming and coal gasification method are the major strategies. However, their sustainable development for hydrogen energy will be restricted by large consumption of fossil fuel and CO2 emission. Because of the requirement of high temperature and pressure for the manufacturing technology, both the catalyst and endanger people's health are suffered from the residual CO intermediate produced by this technology. With the advantage of clean, efficient and sustainable, water electrolysis for hydrogen production has aroused people's attention [14-17]. The overall reaction of water splitting, is constituted by cathodic hydrogen evolution reaction and anodic oxygen evolution reaction as shown in Fig. 1. Benefited by high faradic efficiency, almost no side reaction occurs during electrolysis. The emergence of anion exchange membranes (AEMs) allows the use of two different electrolytes in the anode and cathode compartments of the entire electrolytic cell [18-22]. As the most efficient electrolytes for electrocatalytic hydrogen evolution reaction (HER), acidic electrolyte could be used in cathode compartment to provide a high concentration of protons/hydroniums as reactants. And the alkaline electrolytes could be used in the anode compartment because of the lower overpotential of oxygen evolution reaction (OER) in there. With no-carbon based reactant in system, high purity hydrogen can be easily produced by simple gas separating procedure. The only deficiency of water splitting is the high overpotential to drive overall reaction which significantly restrict its industrialized application. As water electrolysis is a strong uphill reaction, water electrolyzers typically operate at a voltage of 1.8–2.0 V, which is much greater than the theoretical voltage of 1.23 V. Therefore, searching for highly active and stable electrocatalyst is the key to decrease the overpotential of HER and OER. Moreover, the ability to fabricating efficient bifunctional catalysts is appealing due to their characteristics such as reduced production costs and simplified experimental setup. The state-of-the-art HER and OER electrocatalysts to date have been Pt-based materials and oxides of Ru or Ir, respectively, but their scarcity and high cost have hampered their widespread application [23, 24]. Therefore, as shown in Fig. 1, reducing the overpotential of HER and OER is essentially to accelerate the efficiency of the interconversion of chemical and electrical energy, which is the fundamental purpose of the catalyst [25-29].

|
Download:
|
| Fig. 1. (a) Hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) of electrocatalytic water splitting and (b) Schematic representation of HER and OER overpotentials. | |
In the last decades, tremendous breakthroughs have been made in catalyst for the electrocatalytic water splitting, and the results are well-summarized by many excellent review articles in this research domain different catalysts are applied to drive HER and OER will complicate manufacture procedure and raise production cost [30-35]. Among the existing carbon materials, especially graphene, exhibit excellent properties in electrochemical water splitting due to their better conductivity, ductility and high surface area [36-38]. Additional new materials, including layered double hydroxides (LDHs) [39-43], have excellent water splitting properties attributed to the unique structure, abundant interlayer electrons and intermediate adsorption and desorption channels; the emerging MOF materials are also of great interest in water splitting owing to their highly ordered structure [44-48]. Researching bifunctional electrode material to catalyze both HER and OER can not only simplify the procedure in practical application but also benefit to realize the industrialization of multiple secondary energy. Recently, a series electrocatalytic material has been developed to decrease the overpotential of HER and OER, and various bifunctional catalyst has been fabricated with catalytic abilities for both HER and OER, such as oxides [49-52], carbides [53-55], sulfides [56-62] and phosphides [63-67].
In this review, the bifunctional catalyst for overall water splitting can be classified into three types. The first type possesses the intrinsic activity for both HER and OER. Reversing the current direction, the reaction on the electrode will be simultaneously altered with retention of catalytic activity. In the perspective of thermodynamics, the reaction rate is largely determined by adsorption free energy change (ΔG) of intermediate calculated from density functional theory (DFT). As proposed in Sabatier principle, the interaction between catalyst and reactant should be neither too strong nor too weak [68, 69]. Hydrogen adsorption free energy (ΔG·H) can serve as a descriptor to evaluate the binding energy of surface adsorbed H [70-73]. Similarly, ΔG·O – ΔG·OH can describe the interaction between catalyst and adsorbed oxygen species [74-78]. Plotting the catalytic activity against adsorption free energy, various catalysts in same kind will distribute as volcano-type. The catalyst located at the summit of volcano plots will exhibit the better HER and OER activity. Therefore, the first type of bifunctional catalyst for overall water splitting should possess appropriate interaction or binding energy for both hydrogen and oxygen intermediate to approach a near neutral ΔG. Meanwhile, the high potential at OER region can provide strong oxidizing condition. This type of catalyst should not only exhibit high intrinsic activity to OER but also retain the chemical constitution unchanged.
The second type of bifunctional catalysts are intrinsically active to HER. Under the oxidation condition during OER, it will be gradually transformed into corresponding oxides, hydroxides and oxyhydroxide to indirectly catalyze water oxidation. Actually, two compounds respectively catalyze the HER and OER. Therefore, it may imprecise to name this kind of catalysts as bifunctional catalyst with the prospective of intrinsic mechanism. Most bifunctional electrocatalysts of sulfides and phosphides are belong to this category. The third type of bifunctional catalysts are generally integrated by two substance with catalytic activity to HER and OER respectively. The particular heterojunction structure between two substances exist variety of interactions, involving strain effect, fermi level change and electronic structure modulation [79-82]. The surface strain effect originates from interatomic compression or stretch within 6 mono-layer. Fermi level change is generated by the contact between two substances with different work function. Electronic structure modulation is generally resulted by ions exchange from the interface.
In this mini-review, we first focus on the mechanism of overall water splitting to understand the intrinsic driving force for electrochemical HER and OER (Fig. 2). Then, a series of electrocatalyst applied to bifunctional water splitting are reviewed. Herein, we first discuss the bifunctional electrocatalyst for water splitting in three types: direct catalysis, indirect catalysis and hybrid catalysis. Next, we introduce the non-noble metal oxides, carbides, sulfides and phosphates used in the water splitting. Subsequently, combined with the design of material structure, we highlight the structure-activity relationship of heterogeneous materials. Lastly, based on the present attainments, the scientific challenges of bifunctional catalysts for electrolysis of water and the potential development directions are proposed and discussed.

|
Download:
|
| Fig. 2. Transition metal-based catalysts for overall water splitting. | |
As early as 1789, Troostwijk and Deiman has built the earliest electrolytic cell powered by an electrostatic generator. They find that the electric discharge would cause gas evolution including two part of hydrogen gas and a part of oxygen gas. The mechanism of water splitting gradually emerged through numerous researches. As illustrated in Fig. 1a the overall electrolytic cell is constituted by electrolyte, anode, cathode and power source. The overall reaction for water splitting can be described in Eqs. 1-5.
Overall reaction:

|
(1) |
In acidic solution:
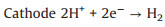
|
(2) |

|
(3) |
In alkaline solution:

|
(4) |

|
(5) |
The positive standard-state free energy change (ΔG0) confirms that the overall reaction should surmount the energy barrier for the formation of H2 and O2. The thermodynamic equilibrium potential for overall water splitting is 1.23 V which contain 0 V for cathodic HER and 1.23 V for anodic OER in 298.15 K and 1 atm. However, practical overall cell voltage (Voc) for water splitting is far from ideal potential, which can be described in Eq. 6:
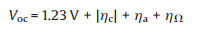
|
(6) |
where ηc and ηa are the overpotentials demanded to drive the cathodic HER and anodic OER, respectively, and ηΩ is the extra potential to compensate the internal resistance loss which mainly consist of solution resistance and contact resistance. What can reduce the operational voltage is to minimize the ηc and ηa originated from thermodynamic and kinetic limitation.
2.1. Hydrogen evolution reactionAs the cathode for water electrolysis and the direct reaction for hydrogen production, HER has comprehensively studied on massive materials. Briefly, HER consists of three steps as illustrated in Eqs. 7-11 [83].
Volmer step (electrochemical adsorption):
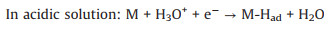
|
(7) |

|
(8) |
Heyrovsky step (electrochemical desorption):

|
(9) |
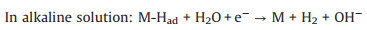
|
(10) |
or Tafel recombination (chemical desorption):
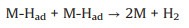
|
(11) |
Firstly, the electrochemical adsorption of hydrogen proceeds on the surface of catalyst, and that is Volmer step. Then, two pathways for desorption of atomic adsorbed hydrogen can be accomplished by electrochemical Heyrovsky step or chemical Tafel recombination. It is clear that the bonding energy to hydrogen should be balanced. The Gibbs free energy change of hydrogen adsorption (ΔGH) can describe the capacity difference between adsorbing and desorbing hydrogen. Plotting ΔGH against catalytic activity, the "volcano plots" can predict the performance of HER catalysts in acid solution [63]. When ΔGH close to zero, most of catalysts exhibit an excellent HER performance. However, rare H3O+ are existed in alkaline solution. The excess water dissociation energy is needed during the formation of M-Had in Volmer step and electrochemical desorption of atomic hydrogen in Heyrovsky step (Eqs. 8 and 10). Therefore, more effort for theorical calculations under alkaline condition will further advance the evaluation system to rationally design HER electrocatalyst.
2.2. Oxygen evolution reactionAs the other side of HER in water splitting, OER has also been extensively explored for various materials. The mechanism of OER is extremely different compared to HER. Four-electron transfer in OER complicate the overall reaction mechanism and make it hard to describe the real reaction process [84]. A general model has been established by Norskov and Rossmeisl et al. based on thermochemistry [85-87]. DFT calculation is utilized to ascertain the thermodynamics of independent reaction steps. This model reaction mechanism as so-called "associative mechanism" is proposed as shown below (Eqs. 12-15):

|
(12) |
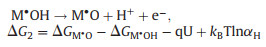
|
(13) |

|
(14) |
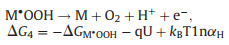
|
(15) |
Firstly, a water molecule is oxidized on the surface of catalyst M to form M·OH and transfer an electron and a proton (Eq. 12). Secondly, the generated MOH is further oxidized to M·O intermediate (Eq. 13). Thirdly, another water molecule is oxidized on M·O intermediate to form a superoxide intermediate M·OOH (Eq. 14). At last, M·OOH intermediate is oxidized to oxygen molecule and desorbed on the surface (Eq. 15). Each reaction step comprises an electron transfer and a proton decoupled indicating that the potential and pH can determine the free energy change (ΔGi, i correspond to reaction step). For notational simplicity, while this OER mechanism is defined under pH 0 at room temperature, kBTlnαH term is equal to zero in Eqs. 12-15. Then, the free energy change is dependent to the Gibbs free adsorption energies of the surface intermediates ΔGM·OH, ΔGM·O, ΔGM·OOH and applied potential U, for instance, ΔG2 = ΔGM·O – ΔGM·OH – qU and ΔG3 = ΔGM·OOH – ΔGM·O – qU. M.T.M. Koper proposed that the difference between ΔGM·OH and ΔGM·OH is 3.2 eV for metal and some metal oxide. Therefore, "ΔGM·O – ΔGM·OH" can serve as a descriptor to evaluate the thermodynamically minimum favorable step in the OER. From the volcano plot summarized by previous authors demonstrates that catalysts with outstanding activity for OER are located around the summit of plots [88]. In another words, an excellent OER catalyst should possess appropriate bonding strength for oxygen intermediate.
2.3. Electrochemical perspective for water splittingElectrocatalysis for water splitting can be ordinarily described as the capability of an electrocatalyst to promote the hydrogen reduction on cathode or oxide oxidation on anode. The key parameters for activity evaluation include the overpotential, Tafel slope, turnover frequency (TOF), faradic efficiency and stability.
2.3.1. OverpotentialDepending on the Nernst equation, the Nernst potentials of HER and OER for the standard conditions (at 25 ℃ and 1 atm) with reference to the normal hydrogen electrode (NHE) are zero and 1.23 V, respectively. However, the reality of the HER and OER process requires a higher application potential to overcome some unfavorable problems, such as kinetic barriers caused by high activation energy and low energy efficiency. Excess potentials (overpotentials, η) are used primarily to overcome the inherent activation barriers present on the anode (ηa) and cathode (ηc), alongside a number of other resistances (ηother), such as solution and contact resistances. Therefore, internal resistance (Rs), due to the internal resistance of the electrocatalyst, the solvent resistance and the contact resistance of the electrochemical system, wires, equipment, etc., will inevitably lead to a Rs and thus introduce an ohmic potential drop, which usually requires correction/compensation of the potential-current density (E - j). A potential required to drive the electrolyzed water is therefore further expressed as: E = E0 + iR + η, where iR is the ohmic potential drop of the current flow and the E0 is the measured potential.
2.3.2. Tafel slopeTafel plots can provide the methods for quantitative and mechanistic characterization for multistep electrochemical reaction. The Tafel slope is recognized as a sensitive parameter for evaluating the electrode polarization rate logarithmically to the current density, describing the dependence of the steady-state current density on various overpotentials. The overpotential (η) is logarithmically related to the current density (j), and the linear part of the Tafel plot accords with the Tafel equation: η = a + blogj; where b is the Tafel slope. Two important parameters can be deduced from the Tafel equation, namely, the Tafel slope (b) and the exchange current density (j0). The Tafel slope (b) is an intrinsic property of the catalyst and is closely related to the rate of the catalyst. Smaller b implies that less overpotential is required to provide the same current density increment, indicating faster electron transfer kinetics. Another key kinetic parameter describing the intrinsic catalytic activity of the electrocatalyst under reversible conditions isj0, which is assumed to be determined when the overpotential is zero. A higher j0 and a smaller b than is required for a good catalyst should be characteristic.
2.3.3. Turnover frequencyThe turnover frequency (TOF) is defined as the number of desired resultants generated per catalytic site per second, which can reveal the intrinsic activity of specific catalytic site. Commonly, the value of TOF can be calculated by: TOF = jA/4nF [89, 90]. The j is the current density at given overpotential in linear sweep voltammetry (LSV) curve; A is the area of the working electrode; n represent the number of moles of the active materials that is calculated using the electrochemically active surface area (ECSA) of the catalyst in the reaction; F are the Avogadro's constant and Faraday's constant, respectively. The TOF value can be calculated with the certain crystalline structure and uniform surface. However, it is difficult to obtain the exact value of TOF due to the complexity of surface, molecule structure and reaction mechanism. Nonetheless, TOF can serve as an important parameter for comparing the activity of homologous catalyst.
2.3.4. Faradic efficiencyFaradic efficiency is a key parameter to reflect electron utilization efficiency during electrochemical process. Regarding to water splitting, the Faradic efficiency can be obtained by measuring the ratio of gas produced experimentally and theoretically. To be precise, the generated gas is collected under a constant current and further measured by gas chromatography. Generally, water splitting has high Faradic efficiency due to the rare side reaction, which benefit to practical application for energy utilization efficiency.
2.3.5. StabilityThe practical application of electrocatalysts for water splitting are typically challenged by the structure and phase stability. The overall water splitting is in two extremes that HER and OER are operated under strongly reductive and oxidative environment, respectively. To exam the stability for water splitting, one method is to perform chronoamperometry in constant potential or chronopotentiometry in constant current density with a long period of time. Generally, the current density or current density of onset potential should larger than 10 mA/cm2, and the duration time is preferably beyond 10 h or longer to approach the industrialized application. Another method is to perform a multi-cycle cyclic voltammetry (CV) or LSV measurement. The number of cycles should beyond 5000 times to measure the stability of electrocatalyst under dynamic variation.
3. Non-noble metal electrocatalysts for overall water splittingThe recently reported works related to overall water splitting are mainly about the transition metal compounds, such as Fe-, Co-, Ni-, and Mo-based materials due to their high catalytic activity in theory, earth abundant and low price. Various these kinds of catalysts reveal high activity to both HER and OER, and the overpotential for overall water splitting even lower to the benchmark Pt/C and IrO2 electrode couple. Ordinarily, the majority of reported bifunctional catalysts are these transition metal oxides, carbides, phosphides and sulfides. The highly catalytic activity is also benefited from the elaborately morphologic and electronic structure modification. As mentioned above, we categorized these bifunctional catalysts in their intrinsic catalytic mechanism, namely, direct catalysis, indirect catalysis and hybrid catalysis. The following section will review the recent advanced progress in structural design, electrochemical performance and catalytic mechanism for overall water splitting in alkaline solution.
3.1. Direct catalysts 3.1.1. Transition metal oxidesOwing to the low cost, high abundance, and considerable anticorrosion properties in an alkaline environment, transition metal oxides were widely developed as OER catalysts. For example, spinel-type oxides (AB2O4, A and B for 3d transition metals such as Ni, Fe, Co, Cu, Zn and Mn) are one of a class of oxidation species that have received a lot of attention [91-94]. Our group has a series of work on the application of spinel oxides to water oxidation, pioneering the research on electrolytic water [95-99]. Co-based oxides are common candidates for high-efficiency OER electrocatalysts such as Co3O4 due to their mixed valence of Co metals, abundant reserves, low cost-effectiveness and high stability [95, 100-103]. However, the electrocatalytic activity is low due to its perfect crystal structure and poor electrical conductivity. Therefore, improving the performance of Co3O4 generally requires optimizing its low conductivity to increase its electron exchange rate. Wang et al. synthesized cobalt oxide/N-doped carbon sheet hybridization (CoOx @CN) via a facile one-pot annealing method by combining cobalt oxide with the conductively superior carbon. Attributed to the excellent electrical conductivity of the carbon sheet and the synergy between Co metal and Co oxide and the electron-donating effects for nitrogen heteroatom doping, the catalyst has improved HER and OER performance and requires only 1.55 V battery voltage when assembled to drive 20 mA/cm2 current density as an overall water splitting electrode [104]. Likewise, Zhang and co-worker enhanced the weak conductivity of the Co3O4 nanorods by designing structural arrays of the nanorods to shorten the electron transfer path and introduce O vacancies, resulting in better electrolytic water catalytic activity [105].
Defect engineering as an effective strategy to regulate the physical and chemical properties of electrocatalysts can achieve higher performance catalytic activity. Wang et al. have developed an efficient Co3O4 based electrocatalyst with oxygen rich vacancies and high specific surface area through a one-step plasma etching strategy [106]. The similar tactic has also been employed in other oxides, where for example, Qiao's group designed a NiO nanorod array with a large number of O-vacancies generated by a facile cation exchange method on a high conductivity CFP [107]. In addition, to combine the introduction of defects and the improvement of electrical conductivity as two ways to improve catalyst performance, Wang et al. successfully prepared Co3O4 nanosheets by a one-step room temperature CH4 plasma, and the obtained Co3O4 has a significant boost in overall water solubilization activity caused by defect enrichment and carbon doping [108]. The large number of exposed effective active sites for the electrocatalyst is also an essential parameter in defining the catalytic performance. For example, porous nanostructures can provide a larger surface area and more active sites with higher HER and OER activity relative to compact nanostructures. Cui et al. produced porous MoO2 via wet chemical method and annealing treatment, yielding a cell voltage of only 1.53 V for current density 10 mA/cm2 and excellent stability when used as a complete water splitting catalyst [109]. These methods are usually based on ion exchange, usually under gas conditions or at high temperatures, to obtain oxygen vacancies and thus promote their catalytic activity. As well, for layered catalysts, the loss of active sites is typically caused due to the layer is too thick to be thoroughly exposed, allowing the catalytic performance to be improved through the preparation of ultra-thin layered materials of atomic level thickness. Zhang et al. have successfully fabricated ultrathin δ-MnO2 nanosheet arrays (NS-MnO2) with a thickness of only two monolayers (1.4 nm) on Ni Foam utilizing a one-step hydrothermalin-situ growth process. The SEM image confirmed that NS-MnO2 consists of a series of 2D MnO2 nanosheets arrayed almost perpendicularly on the NF surface, where the structural advantages of δ-MnO2 favoring exposure to a large number of active sites and associated with mass transfer are clearly demonstrated. And the atomic force microscopy (AFM) revealed that the thickness of the resulting ultrathin nanosheets of NS-MnO2 is about 1.4 nm, which implies the presence of a large number of exposed active sites, favoring the structural advantages of exposure to numerous active sites and correlating with mass transfer was clearly displayed. As a result, the electrode exposes a large number of electrocatalytically active sites and displays excellent electrical conductivity, leading to strong performance against HER and OER. In addition, the DFT calculations reveal the source of NS-MnO2 activity in fully water dissolution, and the adsorption free energy of ·H (ΔG·H) embodies the improved performance of Vo-MnO2 for HER [110]. Therefore, one way to regulate the water electrolytic activation of a sample is to reduce its thickness or size so that more active sites are exposed while generating a large number of vacancies as oxygen vacancies. Also, an elaborately strategy was designed to synthesis coral-like NiFe-based oxide, which contained the approach of generating intermetallic phase Al3Ni and dealloying method. As shown in Fig. 3a, the samples are transformed into mesoporous nanowire network structures after a precursor dealloying operation. And the SEM shows a nanowire bundles-like network structure after dealloying, which is similar to the structure of an integrated coral, this opening network structure facilitates adequate contact with the electrolyte and rapid release of gas (Figs. 3b–d). Thus, benefiting from this unique mesoporous nanowire network structure providing an enhanced specific surface area, accelerated charge/ion transfer and mass transfer, Ni2Fe1-O exhibits a good OER and HER activity and can support current density of 10 mA/cm2 with a cell voltage of only 1.64 V for overall water splitting when used as anode and cathode in an electrolytic cell (Figs. 3e and f) [111].

|
Download:
|
| Fig. 3. (a) Schematic illustration the synthesis route and electrochemical catalytic performance of the Ni-Fe-O mesoporous nanowire. (b, c) SEM images of the coral-like Ni2Fe1-O catalyst with (d) a coral picture. (e) Steady-state polarization curve of an alkaline water electrolyzer using two Ni foam-supported Ni2Fe1-O electrodes. (f) Chronopotentiometry curve of the Ni foam-supported Ni2Fe1-O for overall water splitting and the Photograph. Reproduced with permission [111]. Copyright 2017, Wiley-VCH. | |
It is worth noting that three types of TMOs (NiCo2O4 (NCO), CoMn2O4, and NiMn2O4) with necklace-like multiple hollow structure were first developed with rational synthesis strategies which include simple carbon template-assisted synthesis of complex hollow structures followed by reduction by Peng et al. (Figs. 4a and b) [112]. The R-NCO is a quasi-one-dimensional chain structure of interconnected spheres with the length of a few micrometers, with nanoparticles consisting of triple-shelled hollow spheres with diameters of about 500 nm. Due to the unique necklace-like multi-shell hollow nanostructure and oxygen-rich vacancies, R-NCO exhibited excellent electrocatalytic performance. Of these, an X-ray absorption fine structure spectroscopy (XAFS) analysis by the authors further revealed the apparent oxygen vacancies in R-NCO (Figs. 4c and d). In order to demonstrate the excellent catalytic performance of this catalyst, the authors performed DFT calculations to elucidate the HER and OER catalytic mechanism of R-NCO. The density of states (PDOS) calculations show that it has a low Fermi energy level and thus the ability to reduce the adsorption energy due to the generation of oxygen vacancies from the reduction treatment. For HER, ΔG·H is regularly adopted as a predictor of the theoretical activity of the hydrogen evolutionary reaction in the basic electrolyte, as shown by the reduced ΔG·H of pure NCO shown in Fig. 4e, R-NCO showing the HER performance increase; For OER, a typical four-electron step, the performance increase is demonstrated by the side of the reduced free energy of the rate-determining step shown in Fig. 4f. After a brief understanding of the OER determination steps and improvements to polymetallic oxides, a variety of valuable carbon materials and active substances were designed to enhance OER and HER. For instance, Co-Mn oxides were decorated on a highly ordered carbon by a solvent evaporation-induced self-assembly method and annealing treatment [113]. CoMnO nanoparticles enable optimized OER activity with efficient electronic structure configuration, and the CN framework acts as an excellent HER catalyst. The ordered superlattice structure facilitates enhanced reaction sites, efficient charge transfer and structural stability, and more importantly, doping of pyridine-like N atoms in the carbon framework can significantly increase the reaction sites of HER. In conclusion, the improving catalytic performance of transition metal oxides in electrolytic water generally starts with improving the intrinsic activity or the conductivity of the catalyst, i.e. the electron transfer rate.

|
Download:
|
| Fig. 4. (a) Schematic illustration of the formation process of R-TMO: (I) The absorption of metal ions on the carbon, (II) calcination of the absorbed carbon, and (III) reduction of the TMO to obtain R-TMO with a necklace-like multishelled hollow structure. (b) Schematic illustration of creating oxygen vacancy defects on the surface of NCO after reduction, which is applied as a bifunctional electrocatalyst for water splitting to produce H2 and O2 in 1.0 mol/L KOH aqueous solution. (c) Co K-edge EXAFS data. (d) The corresponding k3–weighted Fourier-transformed data of pristine NCO and R-NCO. (e) Calculated free energy diagram of the HER on pristine NCO and R-NCO. (f) Schematic illustration of reaction paths for OER on pristine NCO and R-NCO equilibrium potential at 1.23 V. Reproduced with permission [104]. Copyright 2018 American Chemical Society. | |
Regarding the poor electrical conductivity and inappropriate H-bonding energy of transition metal oxides, the structural and electronic properties of transition metal carbides (TMCs), examples include, the metallic band state, the tunable surface/bulk structure, the wide range of pH suitability. The inherent superiority of TMCs is highlighted by their high electron conductivity compared to the electrical conductivity of semiconductors of metal sulfides, phosphides and nitrides. More importantly, due to the excellent HER activity and stability, the TMCs are considered to be one of the most promising catalysts in the area of water electrolysis [114-117]. For instance, the intrinsic electrocatalytic activity of molybdenum carbide (Mo2C) for hydrogen adsorption and activation is similar to that of Pt because of its electronic configuration similar to that of noble metals [118-123]. In particular, the Mo2C is an important component of the early TMCs, high crystalline molybdenum carbide is a kind of compound with high melting point. As early as 2012, Hu et al. performed a detailed study of the HER activity of Mo2C under acidic and alkaline conditions [124]. At present, a lot of efforts have been made to study the electrolysis performance of Mo2C, adjust the morphology of the catalyst reasonably to expose more surfaces, obtain ultra-fine nanocrystals to accelerated electron transport. For instance, due to the poor accessibility and agglomeration of pure molybdenum carbides, the high carbonization temperature of the produced carbides affects the improvement of their electrocatalytic activity, so that rational structural design and hybridization of carbonaceous materials can reduce the agglomeration of molybdenum carbide-based materials and greatly improve their performance. Lou's group reported by carburizing Mo-polydopamine nanotubes under a N2 gas flow to synthesize 2D ultrathin nanosheets hierarchical β-Mo2C nanotubes as a superior electrocatalyst for HER (Fig. 5a). It serves as a catalyst for HER and maintains a good catalytic activity under both acidic and basic conditions. A three-step approach is used to rapidly prepare nanorods with core-shell structure, and a large number of vacancies and highly crystalline catalysts are obtained by etching and annealing treatment. Authors claim that the good electrochemical performance can be attributed to: i) Rapid electron transport on high conductivity carbon nanosheet substrates and the unique one-dimensional nanostructure that fully utilize the active site of the catalyst; ii) the highly porous hierarchical hollow structure expands the interface between the electrode and the electrolyte, which is beneficial to the charge and mass transfer in the electrochemical reaction with the strong stability of the catalyst [125]. Likewise, Yuan et al. reports a novel and effective synthetic strategy to immobilize N-doped porous carbon with ultrafine Mo2C nanoparticles (MoC@NCS) under a graphitic carbon nitride (g-C3N4) template by direct carbon thermal reduction of aniline derived polymers and molybdate. As the coupled porous N-doped carbon matrix increases the electrocatalytic surface area of Mo2C, and the interconnected carbon integral optimizes mass transfer and charge channels of the conductive carbon matrix and the strong coupling effect between the N-doped carbon matrix and Mo2C nanoparticles, MoC@ NCS catalyst exhibits good HER performance and stability [126].

|
Download:
|
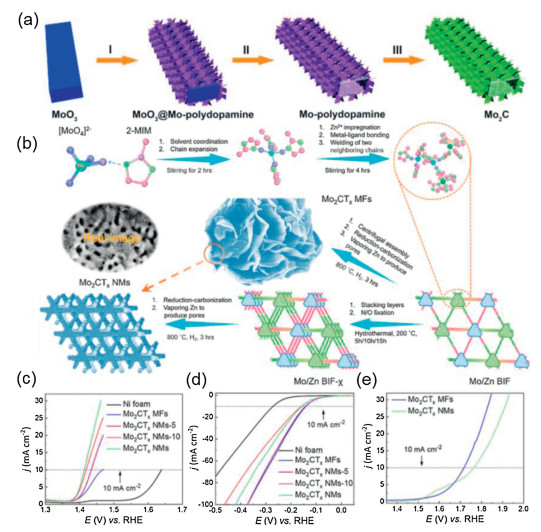
| Fig. 5. (a) Schematic illustration the procedure of hierarchical b-Mo2C nano-tubes. Reproduced with permission [125]. Copyright 2015, Wiley-VCH. (b) Schematic illustration of the synthetic process for Mo2CTx NMs, MFs. LSV curves of Mo2CTx NMs and MFs for (c) OER and (d) HER and (e) overall water splitting. Reproduced with permission [127]. Copyright 2018, Elsevier Ltd. | |
In addition, Wang et al. also reports the 2D nanomeshes and 3D microflowers Mo2CTx, by rationally designing novel Mo/Zn bimetal imidazole frameworks (Mo/Zn BIFs) after pyrolysis (Fig. 5b). The electrocatalysts shows excellent performance in the process of HER, OER and complete hydrolysis. As shown in Figs. 5c and d, the 2D Mo2CTx nanomesh shows a high performance for OER, which can drive a current density of 10 mA/cm2 at a low overpotential of 180 mV, and 3D microflowers Mo2CTx has a better activity for HER, which can reach 10 mA/cm2 at an overpotential of 140 mV. The Mo2CTx MF is also used as a catalyst for overall water splitting and can drive a current density of 10 mA/cm2 at a low voltage of 1.7 V in a two-electrode test system (Fig. 5e). Considering the surface termination of catalytic active nanostructures, the electronic properties of atoms near the surface will change, the Mo LIII-edge X-ray near edge absorption (XANES) study and theoretical calculation show that the surface-terminated oxygen is the key to activate the excellent OER performance, and the Mo atomic position on the surface is the key to activate the HER performance. The results show that surface chemical modification and multi-scale structure design are effective strategies to realize atomic level engineering of active catalysts [127]. As known, Mo2C catalysts are inferior to Pt-based catalysts due to their poor electrical conductivity and slow interfacial reaction kinetics, resulting in slower catalytic activity. Thus, the methods to improve the performance of Mo2C electrolytic water include modifying the morphology to obtain more exposed active sites. Hence, coupling carbon-based supports materials carbon nanotubes (CNTs), graphenes (GRs), reduced graphene oxide (RGO), porous carbon, etc., they probably play an important role: i) Improving the dispersion of nanoparticles avoid agglomeration or sintering; ii) reducing the resistance between active nanocrystals; iii) promoting the transport of electrolytes and H2 and O2 bubbles to reduce the limitation of mass diffusion; iv) affecting the electronic configuration of active sites through interfacial interaction. The TMCs is coupled to the carbon material support substrates by attachment or in-situ growth strategy. As reported by Gao's team, Mo2C nanoparticles is strongly coupled with carbon sheets to make it can foundation as bifunctional catalyst for overall water splitting. Benefiting the synergistic advantages of active sites on the substrate, high surface area and unique layered structure of supporting carbon sheets, the resulting Mo2C@CS shows excellent electrocatalytic activity and excellent stability for both HER and OER. The Mo2C@CS composite can be served as both anode and cathode for the two-electrode water electrolyze, yielding a current density of 10 mA/cm2 at a cell voltage of 1.73 V with robust stability over 100 h [128].
In addition to the direct coupling with carbon materials, the addition of some other elements or electronegative anions in the process of form or self-growth form is also one of the methods to improve the performance. Due to the favorable intermediate adsorption of the optimum OH-Ni2+δ bond strength (0 < δ < 1.5), Ni and its alloy composites have excellent performance of water-splitting in alkaline medium [129-133]. For instance, Yu et al. has reported a novel porous carbon-supported Ni/Mo2C (Ni/Mo2C-PC) composite catalyst obtained via thermal treatment of nickel molybdate nanorods coated with polydopamine, which can efficiently catalysis the HER and OER with remarkable kinetic metrics in alkaline electrolyte. The rod-like structure has a smooth surface with particulate Ni and Mo2C encapsulated within the carbon shells. It is pointed out that the activity of HER is affected by the valence of metal elements, and divalent Ni is an effective hydrolytic active site for the Volmer step in HER reaction. Then, XPS is adopted to correlate the surface composition and enhancement mechanism to obtain electron transfer from Ni to Mo2C, with Mo tending to the lower valence state and Ni to the active Ni2+ valence state, which can improve the performance of HER. This low-cost composite catalyst can efficiently catalyze HER and OER under alkaline solution, and the bifunctional catalyst enables a current density of 10 mA/cm2 at a low cell voltage of merely 1.66 V for overall water splitting. The synergistic effect between Mo2C and Ni nanoparticles and its strong chemical coupling with highly conductive carbon are considered to be the reasons for its outstanding catalytic activity [134]. Similarly, Lu et al. developed a facile electrospinning method coupled with post-carbonation treatment to synthesize nitrogen-doped carbon nanofibers (NCNFs) followed closely to Ni and Mo2C nanoparticles (Ni/Mo2C-NCNFs) as efficient water splitting electrocatalysts (Fig. 6a). Electrospinning technology yields homogeneous nanowire structures that provide good electrical conductivity (Fig. 6b). Thus, the best-matched Ni/Mo2C-CNNFs exhibits the Optimal electrocatalytic activity in 1 mol/L KOH with an overpotential of 143 mV and 288 mV for HER and OER (at j = 10 mA/cm2), respectively. Notably, using Ni/Mo2C-NCNFs as catalysts for the anode and cathode in water electrolyzer, the current density of 10 mA/cm2 split at 1.64 V is impressive with a significant 100 h durability. The mechanism of performance enhancement is as follows: Coupling between Ni and Mo2C provides strong synergy, which are beneficial for active H; doping of N atoms improves the delocalization and electron donor properties of adjacent carbon atoms, increasing the active site and reducing the energy barrier of electrochemical reactions [135].

|
Download:
|
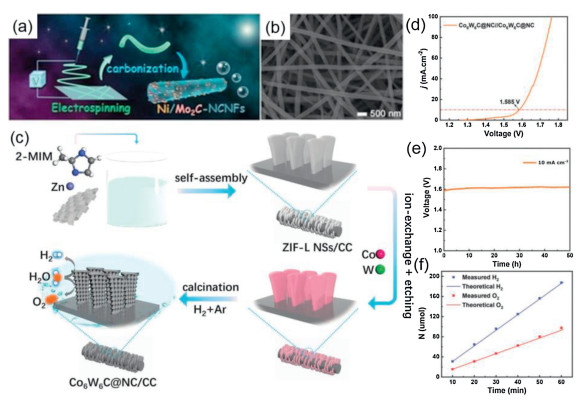
| Fig. 6. (a) Schematic illustration for the fabrication process of Ni/Mo2C-NCNFs nanohybrids. FE-SEM images of the (b) Ni/Mo2C (1:2)-NCNFs. Reproduced with permission [135]. Copyright 2019, Wiley-VCH. (c) The synthesis procedure of Co6W6C@NC/CC. (d) Polarization curves, (e) the long-time chronopotentiometry curve, and (f) faradaic efficiency of overall water splitting. Reproduced with permission [145]. Copyright 2020, Wiley-VCH. | |
Tungsten carbide (WC), analogous to Mo2C, is an abundant and inexpensive catalyst stabilized in acid with a Pt-like catalytic behavior. In 2013, Hashimoto et al. synthesized Fe-WCN electrocatalysts, and they believe that the electron density of W atoms is weakened due to N, thus increasing the catalytic activity of HER [136]. And then, Chen et al. also reported an electrocatalyst incorporating tungsten carbide-nitride and graphene nanosheets, showing that the synergistic effect between W2C and WN phases promotes excellent hydrogen evolution catalysis. The effect, however, cannot be easily fitted to high current densities and the underlying mechanisms remain unclear [137]. To address the fact that WC is capable of catalytic reactions at high current density and to make optimum use of N doping. Sun et al. presented a superhydrophobic catalytic electrode for HER and OER in acids based on N-doped WC nanoarray structures [138]. The authors introduce N-doping to modulate the surface energy level and optimize hydrogen binding, thus promoting her kinetics. And the authors argue that the nano-array structure reveals as well as exposes more active sites for electrochemical reactions, but also, by supplying an underwater ultra-hydrophobic interface, this in return promotes gas release. As a result, the overall water splitting reaction of N-WC nanoarrays as cathode and anode electrodes began at about 1.4 V and the OER showed low overpotential and high activity under acidic conditions. Instead of doping with non-metallic elements (N, S, P), doping with transition metal atoms (e.g., Fe, Co, Ni) regulates surface defects and vacancies, thus affecting the adsorption and dissociation energies of the reactant and product molecules during water splitting [139-144]. More recently, Wang et al. prepares vertically aligned porous cobalt carbide nanosheets embedded with nitrogen-doped carbon matrix (Co6W6C@NC) as efficient overall water splitting catalysts by a facile metal-organic framework (MOF) derivatization method (Fig. 6c). The authors use the characteristics of highly ordered and structurally uniform metal ions in MOFs to prepare porous nanosheets with pure phase bimetallic carbides. The Co6W6C@NC/CC demonstrated excellent HER and OER catalytic performance and also can as a two-electrode water splitting device requiring a low cell voltage of 1.585 V at a current density of 10 mA/cm2 with very stability and the almost 100% Faradaic efficiency indicates high catalytic selectivity in HER and OER processes (Figs. 6d–f). The authors summarize the main reasons for the improved performance: The porous nanosheet structure formed on the carbon cloth can expose more active sites to the electrolyte; moreover, the synergistic interaction of Co and W atoms effectively adjusts the electronic structure and optimizes the hydrogen-binding energy; the Co species introduced are capable of converting to oxide/hydroxide in the OER process to enhance its performance as an OER active center and nitrogen-doped carbon substrates as a backbone to enhance electrical conductivity [145]. Additionally, Zhao et al. reports a simple strategy to synthesize low electronegativity vanadium-substituted cobalt carbides (VxCo3-xC), allowing a large number of defects to be present throughout the lattice, thus enhancing the electrocatalyst performance. The V0.28Co2.72C/CNFs exhibits superior electrochemistry for overall water splitting electrodes, a current density of 10 mA/cm2 can be achieved at only 1.47 V cell voltage. Also, DFT calculations are employed to illustrate that when V replaces Co, the low-electro-negativity vanadium substitution can play a role in weakening the OH bond and thus reducing the OH adsorption energy to optimize its electrochemical properties [146]. Finally, we provide a detailed table summary of the overall water splitting performance of the first type of catalysts (Table 1).
|
|
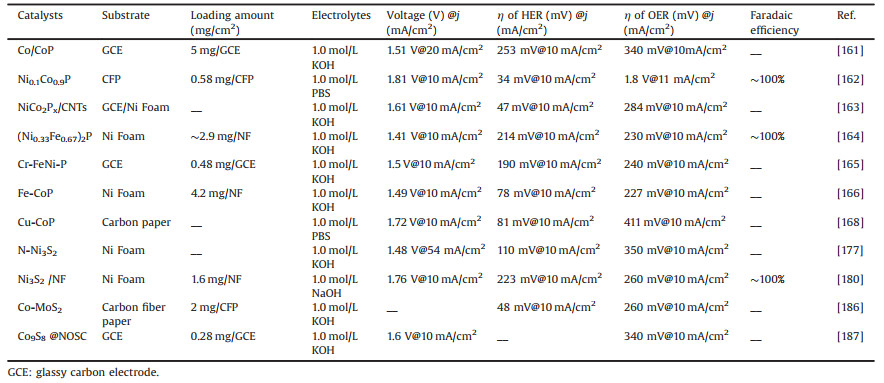
Table 1 A summary of the first type of direct catalysts with electrocatalytic properties. |
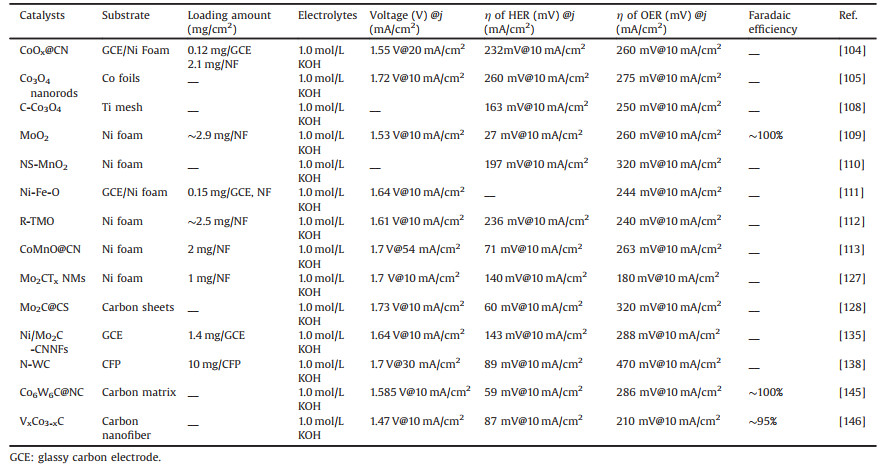
Transition metal oxides and transition metal carbides with intrinsic activity for HER and OER reactions are summarized as the first type of direct catalysts. And the intrinsically active species that change during the OER reaction are classified as secondary catalysts, such as most of the phosphides and sulfides. In 2005, Rodriguez's group performed density functional theory (DFT) calculation to demonstrate that the (001) surface of Ni2P possesses the favorable H binding in hydrogenase systems [147]. And the new strategy for synthesizing nanostructures of transition metal phosphides was described in 2007. A variety of 3d, 4d, and 5d transition metal phosphide nanocrystals (Ni2P, PtP2, Rh2P, Au2P3, Pd5P2, and PdP2) is demonstrated, can be accessed by the solution-mediated reaction of preformed metal nanoparticles. This is particularly important because robust methods exist for controlling the size and shape of single-metal nanoparticles, and these can now serve as reactive templates for the formation of metal phosphides [148]. The hollow morphology is the result of a nanoscale Kirkendall effect, which often occurs for metal phosphide nanoparticles that have been synthesized by reaction of the metal nanoparticle templates. In 2014, CoP nanoparticles are reported to have favorable performance towards HER with only −85 mV overpotentials to attain current density of 20 mA/cm2 in 0.5 mol/L H2SO4, indicating that CoP is an excellent catalyst for HER under acidic conditions [149].
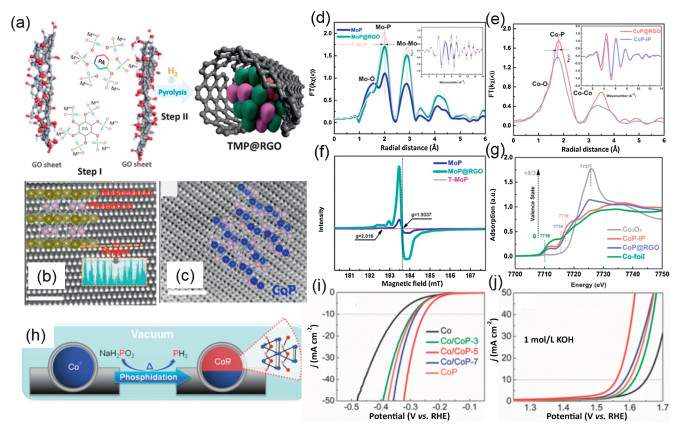
The M–P bond formation in TMPs (M = Mo, Ni, Co, Fe, etc.) seems to have a negligible effect on the electronic properties of the metal species due to the dilution of the M concentration by the presence of P atoms [147]. Moreover, a weak "coordination effect" is induced by the M–P bond, which allows the dissociation of molecular hydrogen with high activity, but also provides moderate binding, captures catalytic intermediates, and prevents catalyst deactivation [150]. Upon occupation of a metal site, the adsorbent (H+/H2O) prefers to interact with the M–P bridge site, where electron transfer to the bridging proton is identified as the rate determination step (RDS) of the entire HER process. Li and co-workers have experimentally reported a universal and scalable method to prepare highly active TMP electrocatalysts for overall water splitting by pyrolyzing phytic acid cross-linked metal complexes (Fig. 7a). High-resolution high-angle annular dark field (HAADF) images of MoP reveal the presence of O atoms (dark spots) in MoP, and HAADF images of CoP show the presence of [001] oriented orthorhombic rhombic bands and an extremely low amount of oxygen (Figs. 7b and c). In order to understand the cause of its good catalytic activity generation, the light absorption around the atom was investigated by extended x-ray absorption fine structure spectroscopy (EXAFS) measurements (Mo, Co). Figs. 7d and e show that Mo and Co atoms are arranged differently with changes in their Mo-P, Co-P and Co-Co bond lengths; it is showed by EPR that not only Mo(III) is present in T-MoP, but that the presence of oxygen makes the Co species abundant in the low-valent state existence (Figs. 7f and g). Moderate introduction of O atoms into the TMPs enhance their intrinsic conductivities and elongate the M-P bonds, facilitating electron transfer to active sites for catalyzing the HER or OER [151]. Although the TMPs have been reported to have high activity for catalyzing HER, yet their active site and stability are not well-understood [152-155]. A new insight into the active sites and stability of CoP nanoparticles (NPs) for HER are provided through experiments and DFT calculations. Research find that the HER activity of CoP decreases as the potential remained above 0.4 V with RHE. This loss of activity is associated with leaching of P and a decrease in the Co/P ratio indicated by the energy dispersive spectroscopy (EDX) results. The formation of (oxy)phosphate(s) coincided with a decrease in activity as confirmed by the PO4 (P5−) signal at the P-K edge X-ray absorption (XAS) and in situ Co-K edge EXAFS. In addition, DFT calculations suggest that surface P can act as a HER active site on CoP, while P-K edge XAS suggests that it may be a P3- species, in contrast to the conventional HER mechanism on metal sites. As such, further research is needed to provide atomic insights into the active site [156].

|
Download:
|
| Fig. 7. (a) Synthesis Process for TMP@RGO by pyrolyzing phytic acid cross-linked complexes high-resolution high-angle annular dark field-STEM (HAADF-STEM) image of (b) MoP and (c) CoP with the corresponding crystal structure superimposed. Mo (d) and Co (e) K-edge extended XAFS oscillation. (f) Room temperature (273 K) EPR measurements for various MoP catalysts. (g) X-ray absorption near-edge structure (XANES) analysis of Co-based compounds. Reproduced with permission [151]. Copyright 2016, American Chemical Society. (h) Synthetic process, and iR-corrected LSV curves for (i) OER and (j) HER. Reproduced with permission [161]. Copyright 2017, Wiley-VCH. | |
Janus nanoparticles with asymmetric shape or composition have different chemical and physical properties due to their broken symmetry. Based on this property, catalysts of the Mott-Schottky class, an example, already apply to dehydrogenation, artificial photosynthesis and other organic reactions [157-160]. The use of their highly coupled interface provides a general principle for the design of catalysts. As shown inFig. 7h, the overall synthesis process for the carbon coated Janus CoP/Co nanoparticles is illustrated. The redistribution of electrons at the Co/CoP interface generally promote the HER and OER activity, greatly extending their applications as highly efficient and stable electrode materials in water splitting devices (Figs. 7i and j). This study opens up a new path to explore Mott–Schottky electrocatalysts made of transition metal phosphides for water splitting reaction, fuel cells, and organic synthesis [161]. Similarly, Yu et al. designs a novel ternary Ni0.1Co0.9P porous nanosheets anchored on conductive carbon fiber paper as a bifunctional catalytic material to efficiently drive water reduction and oxidation reactions in a neutral pH electrolyte. The common CoP crystals are orthorhombic B 31 structures with the formal valence state of Co3+P3− and their crystal domains can be divided into the eg state (directed to the P anion) and the t2g state (involved in the Co-Co bond). The authors' doping of Ni into the CoP lattice leads to a slight compression of the lattice, allowing the tuning of the surface electronic structure to improve the catalytic performance [162].
Establishing an interface with conductive substrates (e.g., NF, copper foam, carbon cloth, stainless steel mesh) with a strong coupling effect enables the electronic structure to be regulated, while improving the electron transfer efficiency and thus the catalytic performance. Our group has reported a facile method to synthesize self-assembled 3D laminar NiCo2Px anchored on functionalized carbon nanotubes (NiCo2Px/CNTs) is developed for achieving cost-efficient, highly-active, and robust electrocatalysts towards overall water splitting in 1 mol/L KOH. Notably, the overpotentials at a current density of 10 mA/cm2 are as low as 47 mV and 284 mV for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), respectively. The water electrolyzer device using NiCo2Px/CNTs as both the cathode and anode only requires a cell voltage of 1.61 V to reach a current density of 10 mA/cm2 along with outstanding durability for at least 48 h [163]. Due to the high adhesion of the generated air bubbles to the powder catalyst, the gas pathway is poor, while the three-dimensional self-supporting structure facilitates an increase in the electrochemically active surface area without the aid of polymer binders, promotes electrolyte penetration, and facilitates electron transport and gas release. Sun's team prepares three-dimensional self-loaded Fe-doped Ni2P nanosheet arrays on Ni foam using a hydrothermal method and in situ phosphorylation as a bifunctional electrocatalyst for overall water splitting (Fig. 8a). The optimized Fe doping of Ni2P (Ni0.33Fe0.67)2P exhibits excellent HER activity-overpotential of ~214 mV to 50 mA/cm2 and enhanced OER activity-overpotential of ~230 mV to 50 mA/cm2 [164]. And as shown inFig. 8b, Wu et al. designs a novel Cr-doped FeNi-P/NCN nanocomposite via a one-step heating treatment using transition metal oxides, dicyandiamide (DCDA) and (NH4)3PO4 as precursors. The superior catalytic performance of the samples is attributed to the fast mass/electron transfer and easier gas release due to the strong interaction between Cr-doped FeNi-P and NCN. What's more, in Fig. 8c the current density of 10 mA/cm2 is achieved with only 1.5 V cell voltage for overall water splitting [165]. Next, Lu et al. proceeded by directly growing Co-Fe Prussian blue analogues on three-dimensional porous conducting substrates, which further phosphorylated into bifunctional Fe-doped CoP (Fe-CoP) electrocatalysts. The prepared Fe-CoP/NF electrode exhibits high electrocatalytic performance and reliability for both OER and HER, can provide a current density of 10 mA/cm2 in 1.0 mol/L KOH solution with a cell voltage of only 1.49 V for overall water splitting [166]. Hu and coworker report a bimetallic (FexNi1−x)2P nanoarrays as efficient electrocatalysts for oxygen evolution in alkaline and neutral media. Benefiting from the optimized Fe doping to adjust the available active surface site and electronic structure, the catalyst can act as an efficient electrocatalyst through the heterogeneous structural interface formed in situ, the porous nanosheet array and the highly open three-dimensional hierarchy [167]. A neutral water electrolytic catalytic reaction can use ocean, lake, or river water directly without the need for expensive, environmentally friendly membranes or separators, and can reduce the cost of electrochemical systems, as opposed to acidic and alkaline conditions. Recently, the ternary Ni0.1Co0.9P porous nanosheet structure proposed by Yu et al. previously mentioned can drive a current density of 10 mA/cm2 at 1.89 V under neutral conditions. However, the problem is still the need for higher cell voltages due to the higher overpotential of the bifunctional electrocatalyst under neutral conditions [162]. Shen et al. reports a facile and controlled method to tailor the electronic structure by Cu doping and developed a highly active electrocatalyst consisting of an array of CoP nanosheets on carbon paper. Owing to its excellent conductivity and optimized electronic structure, the Cu-CoP NAs/CP can drive current density of 10 mA/cm2 with a low overpotential of 81 mV and 411 mV for HER and OER. To understand the material changes that occur during the catalytic process, the authors use XPS analysis to obtain the conversion of Co to a hydroxyl oxide, confirming its intrinsic active species. And DFT calculations also proved by the authors that doping with Cu elements can significantly improve the conductivity of CoP as well as modulate the electronic structure to facilitate fast charge transfer and optimize the adsorption free energy of reaction intermediates (Figs. 8d and e) [168].

|
Download:
|
| Fig. 8. (a) Schematic illustration of the synthesis process of Fe-doped Ni2P nanosheet arrays. Reproduced with permission [164]. Copyright 2017, Wiley-VCH. (b) Schematic of the fabrication of the Cr-doped FeNi–P/NCN, (c) LSV curve of overall water splitting. Reproduced with permission [165]. Copyright 2019, Wiley-VCH. (d) HER free energy diagrams for various sites on pristine and Cu-CoP (111) surfaces, (e) Free energy diagrams at 0 V on the CoOOH and Cu-CoOOH surfaces. Reproduced with permission [168]. Copyright 2020, Elsevier Ltd. | |
So far, transition metal sulfides in the catalytic reaction of HER and OER possess a high activity may be due to their special electronic properties, high valence, synergy between transition metal ions, and strong corrosion resistance in an alkaline environment [169-171]. Ni3S2 is a naturally occurring metal-sulfur group compound that is well suited for various electrochemical applications due to its high electrical conductivity and low cost [172-174]. The Sun's team has already reported the use of amorphous Ni-S films prepared by a simple electrodeposition method as an efficient, robust and inexpensive HER catalyst in a variety of aqueous media including strongly acidic, neutral, strongly alkaline and natural water [175]. Considering the inherent lack of conductivity of fluorine-doped tin oxide (FTO), the use of a more conductive NF as a substrate is a very good choice. For this purpose, Zhang et al. use a simple hydrothermal reaction to successfully decorate single-crystal Ni3S2 nanorods on NF as an excellent OER electrocatalyst. The OER catalytic performance is well improved due to the synergistic chemical coupling effect between hydrated Ni3S2 nanorods, Ni oxide layers and Ni foam support [176]. And Xie et al. highlight a new N-anion-decorated metallic Ni3S2 electrode material with a three-dimensional electrode that has excellent catalytic activity for both HER and OER processes under alkaline conditions. And the authors claim N anion introduction remarkably modifies the morphology and electronic structure of Ni3S2, bringing high exposure of surface active sites, enhanced electrical conductivity, optimal HER Gibbs free energy (ΔG·H) and water adsorption energy changes (ΔG·H2O) [177]. Rationally designed and synthesized the active sites via sizing and assembly modes for trimming nanocatalysts is also a way can be to significantly improve the electrocatalytic performance, but increasing the crystalline density of the most catalytically active surfaces on various nanomaterials is most promising to for their water splitting catalytic performance close to or even better than that of precious metals [178, 179]. Zou and coworker first report a method for synthesizing high exponential plane Ni3S2 nanosheet arrays by in situ growth on foam nickel (NF) [180]. Two sets of lattice fringes of Ni3S2 are revealed in HRTEM, and the angle of entanglement between the (021) and (003) faces of 70.7° is very close to the theoretical value of 70.8° which can indicate that the exposed facets of the nanosheets are. Experimental results and theoretical calculations indicate that Ni3S2/NF's excellent catalytic activity is mainly due to the synergistic catalytic effects produced in it by its nanosheet arrays and exposed (210) high-index facets. Modulation of the effective active site is a strategy to improve the electrocatalytic performance. Recently, Li et al. demonstrate that ammonia treatment is an effective strategy to simultaneously enrich the active sites on MoS2 nanosheets and enhance their intrinsic catalytic activity against HER in basic electrolytes. Through experimental data and theoretical analysis, it is evident that the enriched active MoS2 has long been a focus in electrolytic water hydrolysis. It is known that the application of 2H-MoS2 in catalytic HER is limited by density, active site reactivity, poor charge transport performance between 2H-MoS2 layers and low electrical contact efficiency with conductive carriers [181, 182]. Alternatively, the metal 1T phase MoS2 (1T-MoS2) exhibits easier charge transfer characteristics and is therefore able to expose more active sites, resulting in better performance [183, 184]. The performance can be further improved by engineering the electronic structure of the catalytic site at the atomic level and improving atomic utilization. Zhao et al. reported for the first time that the cobalt covalently doped MoS2 was induced by a covalent doping method for overall water splitting bifunctionality, and the obtained cobalt covalently doped MoS2 showed excellent bifunctional catalytic properties for HER and OER with starting potentials of −0.02 and 1.45 V, respectively, in site with favorable H adsorption free energy is responsible for the good catalytic activity of N-doped Ni3S2 [185]. DFT calculations demonstrate a stable material structure with a Mo/Co ratio of 8:1, and the density of states (DOS) of Co 3d and S 3p show the overlap of its neighboring S atoms in MoS2, confirming the formation of Co-S covalent bonds (Figs. 9a–c). Moreover, Gu's team proposed a reliable and tunable synthesis strategy based on a polymetallic oxygenate template with an Anderson-type polymetallic oxygenate as a precursor to atomically engineered metal doping sites on metal 1T-MoS2 [186]. The outstanding HER activity exhibited by NiO@1T-MoS2/CFP (T-180) is due to the precise co-doping of Ni and O atoms into ultra-thin 1T-MoS2 nanosheets, effectively reducing the kinetic barrier required for the initial dissociation step while facilitating the intermediate state hydrogen production process.

|
Download:
|
| Fig. 9. (a) DFT calculated vapor-phase hydrothermal (VPH) transformation reaction energy of β-Mo2C to 2H-MoS2. (b) The side and top views of the optimized stable structure of Co covalently doped MoS2 with a Mo/Co ratio of 8:1 (green, yellow, and blue balls represent Mo, S, and Co, respectively). (c) The calculated DOS of 3d Co and 3p S states. Reproduced with permission [185]. Copyright 2018, Wiley-VCH. (d) Synthetic procedures used to synthesize Co9S8@NOSC hybrid electrocatalysts; performance of an overall water electrolyzer involving as electro-catalyst. (e) Polarization curves. (f) Chronopotentiometric (i–t) curves. Inset in (f) shows the amounts of theoretically calculated and experimentally determined O2 and H2. Reproduced with permission [187]. Copyright 2018, Wiley-VCH. | |
Based on the above optimization model, the metallic properties of the co-doped MoS2 plates are further revealed, contributing to the electrocatalytic boost. In addition, in combination with the covalent doping of metals, co-doping of non-metallic elements such as N, O, S and other electro-negative elements is also an effective means to improve the performance of electrolytic water. Wu et al. present a facile synthesis of nitrogen, oxygen, and sulfur tri-doped carbon-encapsulated Co9S8 nanoparticles (Fig. 9d) [187]. Thanks to the synergistic effect between the Co9S8 nanoparticle nuclei and the heteroatom-doped carbon shell layer of the material, the Co9S8@NOSC catalyst exhibits a good OER performance and can be applied to overall water splitting (Figs. 9e and f). A two electrode electrolyzer is assembled with Co9S8 @NOSC as an electrocatalyst with current densities of 10 and 20 mA/cm2 at potentials of 1.60 and 1.74 V, respectively (Table 2).
|
|
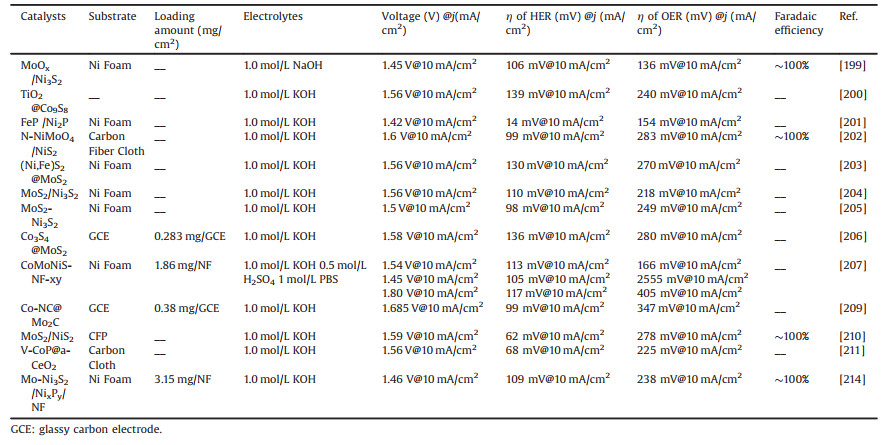
Table 2 A summary of the second type of indirect catalysts with electrocatalytic properties. |
The second type of catalysts mainly have intrinsic catalytic activity for HER, while for OER process their intrinsic catalytically active substances will be transformed (hydroxides, hydroxyl oxides, etc.), and recent papers mainly compare the transformation of their intrinsically catalytically active substances by XPS characterization before and after the reaction, which can be further analyzed by in situ X-ray absorption spectroscopy in more detail. To better compare the differences between the catalysts, we summarized the performance of the second type of catalysts used in this review for overall water splitting as shown in Table 2.
3.3. Hybrid catalysisThe first type of catalysts has an intrinsic catalytic activity towards OER and HER, according to this review, and the method to optimize the activity focuses on regulating the electronic structure, increasing the lower conductivity, and accelerating the electron transfer rate. The second category of catalysts is defined as "indirect catalysts" due to the fact that in the OER process, most catalysts are intermediately converted to substances that have intrinsic catalytic activity for OER activity, as hydroxides. So as the third type of catalyst, on the other hand, is summarized as a heterogeneous catalyst, which usually consists of a combination of two catalysts, such as some transition metal catalysts that are held together by heterogeneous junctions, and they are usually able to combine the advantages of both to meet the requirements of being a bifunctional catalyst. There is a consensus that the strong coupling interface between HER and OER active components usually significantly promotes the final performance of these hybrid electrocatalysts. Due to the possible synergistic effect and the improvement of electron transfer efficiency at the interfaces of different components, the strong coupling interface between HER and OER active components usually significantly promotes the final performance of these hybrid electrocatalysts [188-190]. Simultaneously, interface design has attracted a tremendous quantity of research as one of the most critical and prospective drivers for boosting electrochemical reactions [191-197].
The application of heazlewoodite-Ni3S2 in electrolytic water has been described in the previous section, considering that it does not catalyze both HER and OER efficiently [198]. To this end, Zou's team reports for the first time a synthetic approach that yielded an efficient and ultra-stable electrocatalyst based on Ni3S2 for the overall water splitting. The material is assisted by a small amount of ammonium molybdate to form a unique ultrathin nanosheet structure with hollow Ni3S2 microspheres supporting the formation of a unique structure in a NF material (Fig. 10a). A small amount of MoOx was formed in the reaction, which in combination with Ni3S2 is referred to as MoOx/Ni3S2/NF. The authors experimentally prove the materials possess strong electrocatalytic activity for both HER and OER due to its high density of active exposure sites and fast electron transport channels. In an alkaline electrolysis cell with a current density of 10 mA/cm2 and an applied potential of 1.45 V, the material has a significant stability (> 100 h) [199]. Similarly, leveraging the strengths of structural engineering, Tu et al. coupling the material to the hollow TiO2. Benefited from a larger active surface area, richer/shorter ion/electron transfer channels, and stronger structural stability, and the obtained TiO2@Co9S8 core-branch array exhibited superior electrocatalytic performance (Figs. 10b–d) [200]. The work above is mainly improving the electrochemical performance via structural engineering andinterfacial processes that increase the effective active area of the catalyst along with the electron transfer rate. Ren et al. reported an efficient hybrid catalyst constructed of iron and dinickel phosphide on NF to drive the HER and OER. Surprisingly, the FeP/Ni2P catalysts drive 10 mA/cm2 current only need 1.42 V with an outstanding durability delivers 500 mA/cm2 at 1.72 V over 40 h for overall water splitting. To better understand the catalytic mechanism, the authors confirm the FeP hybridized atop with Ni2P results in a significant increase of the ΔGH by DFT calculations. Also, the authors highlight that the calculations are limited to the thin layer of FeP crystals, ignoring the size of the particles (5−30 nm), so this may be a good direction for research [201].

|
Download:
|
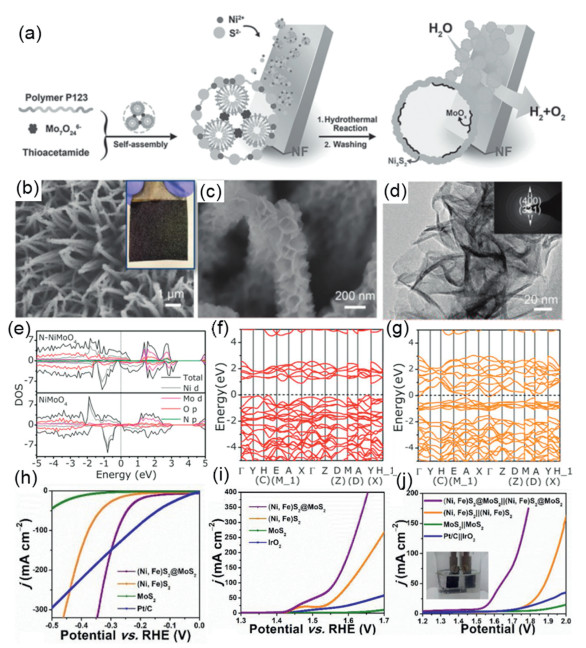
| Fig. 10. (a) Schematic illustration of the synthesis and the structure of MoOx/Ni3S2/NF. Reproduced with permission [199]. Copyright 2016, Wiley-VCH. (b, c) SEM images (optical photograph in inset). (d) TEM image (selected area electron diffraction (SAED) pattern in inset). Reproduced with permission [200]. Copyright 2018, Wiley-VCH. (e) Projected density of states of N-NiMoO4 and NiMoO4. (f) Band structure of pristine NiMoO4. (g) Band structure of N-NiMoO4. Reproduced with permission [202]. Copyright 2019, Wiley-VCH. The LSV curves of (h) HER, (i) OER and (j) two-electrode cell assembled by different materials. Reproduced with permission [203]. Copyright 2019, Elsevier Ltd. | |
The excellent performance of the electrocatalyst is related to its geometry, and the introduction of electronegative elements can also regulate the performance of the catalyst. Benefiting from the high flexibility and inherent anisotropy of one-dimensional materials, as well as the large surface area and rich active edges of 2D materials, in addition to the fact that bimetallic nanocrystals with high separation components can generate vacancies or defects to enhance performance. Xi et al. designs the N-NiMoO4/NiS2 nanowire/nanosheet material with N-controlled electronic conditions. The authors adopt a "top-down" strategy–firstly, prepared parental N-NiMoO4 nanowires by regulate nitridation, which activated Ni sites in the NiMoO4 crystal structure, and then prepared N-NiMoO4/NiS2 with a rich epitaxial heterogeneous interface via selective sulfuration. DFT calculations understand the effect of N doping and confirm the charge transfer from N-NiMoO4 to NiS2 across epitaxial heterogeneous interfaces at the atomic level, and the synergy between the N-NiMoO4 and N-NiS2 domains enhances their HER/OER performance in alkaline solutions (Figs. 10e–g). At the same time, the introduction of N allows the higher state near Fermi level to lift and new states to reduce the band gap, enhance the adsorption strength of molecules and free radicals (·H and ·OH) and enhance conductivity. The abundant electrons on NiS2 will stabilize the H adatom by transferring electrons from N-NiMoO4 to NiS2 to favor it is HER catalytic activity [202]. Synergistic effects from polymetallic and interfacial effects from heterogeneous junctions are often the origin of catalyst activity. Shao et al. prepared (Ni, Fe)S2@MoS2 by hydrothermal sulfured process with molybdenum salts using NiFe LDH as precursor, which serves as an efficient, low-cost stable electrocatalyst under interface engineering effects and synergistic effects [203]. In Figs. 10h–j, the prepared (Ni, Fe)S2@MoS2 catalysts exhibit excellent electrochemical activity and durability in an alkaline environment, with low overpotentials of 130 mV and 270 mV for HER and OER to provide a current density of 10 mA/cm2, respectively. At the same time, when used as an overall water splitting catalyst, the electrolytic cell only requires a potential of 1.56 V at 10 mA/cm2, much lower than other catalysts. In addition to applying interface engineering effects, regulating the morphological structure of the catalyst is also a viable tool. Feng et al. for the first time, propose the interfacial engineering of novel MoS 2/Ni3S2 heterogeneous structures prepared on NF. And the authors combine the DFT calculations and experimental results to confirm the interfacial interaction between MoS2 and Ni3S2 and the in situ generation of NiO can promote the synchronous chemical adsorption of hydrogen and oxygen-containing intermediates, enhancing the overall water splitting performance [204]. The aforementioned work carefully analyzes the role of interfacial interactions between the two catalysts for electron transport and for the energy of adsorption of active substances in catalytic reactions. Experimental simplicity is also an important factor, and for this end, Gao and his coworker utilizing anisotropic molybdate intermediates to directly grow MoS2-Ni3S2 heterogeneous nanorods by a simple one-step hydrothermal method. This MoS2-Ni3S2 electrocatalyst showed superior performance for HER and OER, and the battery voltage is only 1.50 V with a battery voltage can reach current density 10 mA/cm2. The excellent catalytic performance of the samples is attributed to the combination of MoS2-Ni3S2 with better adsorption of H- and the O-containing intermediate, the efficient exposure of the active interface in the layered nanostructures, and the fast electron transport brought about by the anchoring to the NF substrate [205].
Besides the usual 1D nanowires combined with 2D nanosheets, fabricating a core-shell structure is also a good option. Guo et al. developed a core-shell Co3S4 @MoS2 heterostructure by performing a two-step heated hydrothermal reaction and thermal annealing process, whose unique hollow and core-shell structure, along with low charge transfer resistance and abundant active sites, bestows efficient catalytic performance [206]. Coincidentally, Mercouri G. Kanatzidis et al. prepare a hierarchical structure of MoS2/Co9S8/Ni3S2/Ni based on NF as a highly efficient catalyst for overall water splitting in a wide range of pH. This work exhibits good overall water splitting performance over a wide pH range due to its unique 3D hierarchical structure and electron transfer due to the synergy between the different components [207]. In terms of structure and morphology, in addition to the hierarchical nanosheets stacked on a one-dimensional substrate, the preparation of porous nano-heterojunctions is also a good method. One typical example is that Wang et al. developing a bottom-up approach is established for facile synthesis of holey 2D transition metal carbide/nitride heterostructure nanosheets (h-TMCN) with regulated hole sizes by controlled thermal annealing of the Mo/Zn bimetallic imidazolate frameworks (Mo/Zn BIFs). The authors reveal that the Mo/Zn BIFs precursors undergo three interrelated transition steps through phase shift and structure identification. And X-ray fine structure analysis and high-resolution X-ray photoelectron spectroscopy reveal the ability of Mo2C and Mo2N to chemically bond together, producing an enriched N-Mo-C active interface facilitating the water dissociation [208]. The robust porous structure and high surface areas of metal-organic framework (MOFs) may give its unique advantages in electrocatalytic reactions (such as HER, OER). Mu's team developed a method based on a simple MOFs to prepare bifunctional Co-NC@ Mo2C composite catalysts. Different from traditional carbon encapsulated functional nanomaterials, carbon-based framework derived carbonitrides and Co nanoparticles (Co-NC) are encapsulated by Mo2C. With to the unique reverse-encapsulation structure and the synergistic effect of Mo2C and Co-NC, the Co-NC@Mo2C complex catalysts shows excellent catalytic performance for HER and OER in a wide pH range. This study provides a new path for the design of MOF derivative catalysts to obtain multi-functional catalytic performance [209].
The application of defect engineering is also a powerful way to improve the performance of electrocatalysis. Independent, defect-rich heterogeneous-MoS2/NiS2 nanosheets are designed in two steps by Feng et al. for overall water splitting (Fig. 11a). The abundant non-homogeneous phase interface in MoS2/NiS2 provides abundant electroactive sites as well as promotes electron transfer to further synergize the electrocatalytic reaction, thus exhibiting enhanced electrocatalytic performance [210]. Regulating the electronic structure through non-metallic elements is an effective means, from which it is conjectured that the introduction of heterogeneous metals into transition metal materials is also a promising strategy to stimulate intrinsic catalytic activity by modulating electronic structure and morphology. Recently, Jiao et al. synthesize a layered heterogeneous nanoarray composite of V-CoP@a-CoO2 as an ideal and stable electrocatalyst for water splitting. Predictably, as shown in Figs. 11b–d, V-CoP@a-CeO2 exhibits superior performance in both hydrogen evolution and oxygen evolution reactions, and furthermore, the combination of the two-electrode electrolytic cell with the V-CoP@a-CeO2 electrocatalyst requires only an electrolytic cell voltage of 1.56 V and 1.71 V to achieve 10 and 100 mA/cm2 current density, respectively. In the Figs. 11e and f, DFT calculations demonstrate that the V-CoP@a-CoO2 can be used as an excellent electrocatalyst for overall water splitting under alkaline conditions, using the electron transfer exchange rearrangement produced via the doping of V and between CeO2 with V-CoP heterogeneous components [211]. Also, the introduction of Mo and Fe into Ni3S2 bent nanosheets has long been shown to be a method to enhance the performance of the overall water splitting [212, 213]. Mu et al. report the successful synthesis of a novel Mo-doped hollow nanorod structure (Mo-Ni3S2/NixPy/NF) on NF by a simple solvothermal process followed by partial phosphorylation. The electronic structure and activation of Ni sites in Ni3S2 nanostructures are optimized by precise and controlled Mo doping to form Mo-Ni3S2 flower-like nanosheet clusters; subsequently, the formed Mo-Ni3S2 was partially phosphorylated to form hierarchical branched hollow nanorods Mo-Ni3S2/NixPy/NF, resulting in abundant heterogeneous interfaces. The authors also demonstrate experimentally and DFT calculations to showing the presence of electron exchange and heterogeneous composition between Mo-Ni3S2/NixPy/NF can optimize the Gibbs adsorption free energy of oxygen-containing intermediates (ΔG·M) and hydrogen (ΔG·H) to improve OER and HER performance [214]. As for the hybrid catalyst, the current research on its mechanism is mainly based on the DFT calculation, indicating the influence of its specific charge transport and distribution on the adsorption energy of the active intermediate, and figuring out the specific effect between the two is of significant assistance to the understanding of the catalyst. As well, a detailed summary table of the water splitting properties of the above catalysts can be found for a clearer overview of the performance characteristics (Table 3).

|
Download:
|
| Fig. 11. (a) Schematic illustration for the formation of defect-rich heterogeneous MoS2/NiS2 nanosheets. Reproduced with permission [210]. Copyright 2019, Wiley-VCH. LSV polarization curves of the samples with iR-corrected (b) HER, (c) OER and (d) overall water splitting. DFT calculations. (e) The mechanism illustrations of the electrocatalytic HER under alkaline conditions and ΔG diagram of samples. (f) Deformation charge density of V-CoP@CeO2. Reproduced with permission [211]. Copyright 2020, Wiley-VCH. | |
|
|
Table 3 A summary of this third type of hybrid catalysts with electrocatalytic properties. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The development of inexpensive, efficient and stable bifunction catalysts (HER/OER) is of critical importance in water separation technology for hydrogen energy. This minireview describes the application of non-noble transition metal-based electrocatalysts to overall water splitting, which are broadly classified into three types of catalysts based on the intrinsic active site of the electrolytic water reaction process. Considerable efforts have been made to increase the number of active sites (morphological structure regulation, creating material to be porous structures, integrating with conductive carbon supports, surface defects, etc.) and to regulate the electronic structure (heteroatomic doping, vacancies, coupled heterogeneous junction, etc.) to enhance the intrinsic activity and to develop non-precious transition metal-based catalysts for electrolytic water applications more effectively. Among the first direct catalysts, for transition metal oxides, the inherent low conductivity, sluggish reaction kinetics, limited adsorption sites for H intermediates, and improper H-binding energy of most TMOs are obstacles to realizing high-efficient electrocatalytic performance. For transition metal carbides, the sluggish OER process is a major impediment to limiting their application in overall water splitting. For the second type of indirect catalysts, due to the high electronegativity of elements such as P and S, the combination with transition metals can withdraw electron density from the metal atoms and act as effective local sites to trap the positively charged proton. For the third type of hybridization catalyst, it is usually coupled to two different catalysts through heterogeneous junction etc., and due to the properties of combining the two has excellent performance for overall water splitting. In spite of significant advances in material design/synthesis and behavior research, challenges remain in the further development of efficient non-precious metal-based bifunctional electrolytic water catalysts.
Firstly, the challenge with transition metal-based catalysts in overall water splitting is that the HER performance in alkali does not meet or exceed the performance of precious metals, however the catalyst's overall water splitting performance is not only determined by the HER performance, but also by the OER. For practical installations and wide applications, a two-electrode arrangement must be developed to ensure that both HER and OER half-reactions can be effectively performed. Although most HER promoters performed better in acidic media and almost all OER catalysts were better adapted to alkaline conditions, under identical experimental conditions, few non-precious metal-based catalysts were effective in catalyzing the HER and OER reactions. Consequently, it is essential to enhance the HER performance of transition metal-based catalysts in alkaline conditions.
Furthermore, one of the important merits of non-precious metal-based catalysts over precious metal catalysts in terms of low cost and abundance of reserves. Several strategies for the preparation of effective transition metal-based overall water splitting catalysts are developed, however, the complex synthesis process and harsh reaction conditions ultimately lead to high product costs, insufficient catalyst yields and poor catalyst quality. In addition, the synthesis and application of method catalysts, which are limited to the laboratory, are still in a laboratory stage and cannot meet the requirements for industrialization and commercialization. As a result, difficulties have always existed in developing mild methods that enable the large-scale production of high-performance transition metal-based catalysts.
Third, the excellent electrical conductivity of the electrocatalyst, enabling excellent electron transfer during the reaction, can be one of the essential factors affecting the good performance of the electrocatalyst. The poor electrical conductivity of transition metal-based catalysts has been a limiting factor in achieving better catalytic performance. Currently, although most of the methods to improve the conductivity of catalysts are to improve the intrinsic conductivity of MoS2 from the 2H phase of a semiconductor to the 1T phase structure of a metal, and the coupling to a conductive substrate including graphene, carbon-based materials (carbon nanotubes, carbon cloth, carbon paper, carbon fiber), and various foam metals (nickel foam, iron foam, copper foam, and foam alloys). Increasing the exposure of large surface-active sites is an effective initiative in the reaction system to shrink the electron transfer routes between the catalytic component and the electrolyte. Generally enlarged exposed surface-active sites can be attained by coupling with the conductive media mentioned above. However, the durability of the catalyst is not maintained in the long term when surrounded by strong acid and strong alkaline electrolyte, probably as a result of the degradation of the active material in the presence of the strong electrolyte due to massive gas emission and corrosion of the support by the strong electrolyte. Accordingly, the development of a catalyst with good electrical conductivity and favorable stability is also one of the directions in which catalytic performance can be improved.
In the process of searching for advanced overall water splitting catalysts, a basic understanding of the mechanism is necessary. For the multiatomic doped or constituted hybrid heterogeneous structures of complicated compounds, elucidating the actual active site of the catalytic mechanism reaction will be a challenge. Despite the fact that DFT calculations are used to predict the reaction intermediates and active sites of catalysts and to design efficient electrocatalysts, the true catalytic mechanism of the multicomponent system is not totally understood under the established theoretical model. And the basic catalytic mechanism of transition metal-based electrocatalysts remains controversial, as the current interpretation is mostly based on the classical theory of precious metal catalysts, and the interpretation of OER reactions is not very fluid. Therefore, we also need in-depth and systematic mechanistic studies at the atomic level, unable to understand most of the developed catalysts, especially the composite ones.
The existing work analyzing changes in catalysts in the reaction process, while analyzing pre- and post-reaction characterization, may lose important information such as the microstructural evolution of the surface atomic layer and the reaction intermediates absorbed by the catalyst surface during the reaction. In situ spectroscopic studies are powerful tools for unraveling the catalytic mechanism and structure evolution of electrocatalysts, generally involving complex surface structure reconstruction under different redox steps. Analytical techniques such as X-ray absorption spectroscopy, Raman spectroscopy for the reaction process have been successfully applied to track the surface species in the catalyst, helping to reveal the true catalytic active site and catalytic mechanism. For the first type of catalysts, due to intrinsic catalytic activity for HER, the corresponding theoretical calculations are more mature, and the material changes before and after the reaction are well understood, but the evolution of the specific process is lacking. In addition, the process of HER reaction lacks a correspondingly detailed in situ characterization technique, and the understanding of the strength of the H-bond remains only at the theoretical stage for noble-metal catalysts, influenced by electrolytes and other conditions in the real progress. Similarly, the second type of catalysts are not well studied for the specific material transformation of OER, and only the catalytic mechanisms such as LDH have been studied completely but are still controversial. The third type of catalyst, due to its hybridization, also limits the DFT calculation to a few atomic layers on the surfaces, which is not well considered for large thicknesses. Therefore, the development of in situ characterization increases our understanding of its operating state and will help us to further understand its catalytic mechanism.
Finally, the increasing demand for electricity in modern society makes the consumption of electricity not negligible in addition to the cost of manufacturing electrocatalysts. The integration of water electrolysis units into other renewable power sources, such as solar photovoltaic systems, wind, geothermal or tidal generators, is also a promising way of proceeding.
Declaration of competing interestThe authors declare no declarations of interest.
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (No. 21875048), Outstanding Youth Project of Guangdong Natural Science Foundation (No. 2020B1515020028), Major Scientific Project of Guangdong University (No. 2017KZDXM059), Yangcheng Scholars Research Project of Guangzhou (No. 201831820), Science and Technology Research Project of Guangzhou (No. 202002010007).
| [1] |
P. De Luna, C. Hahn, D. Higgins, et al., Science 364 (2019) eaav3506. DOI:10.1126/science.aav3506 |
| [2] |
M.S. Dresselhaus, I.L. Thomas, Nature 414 (2001) 332-337. DOI:10.1038/35104599 |
| [3] |
X. Zou, Y. Zhang, Chem. Soc. Rev. 44 (2015) 5148-5180. DOI:10.1039/C4CS00448E |
| [4] |
Y. Hou, X. Zhuang, X. Feng, Small Methods 1 (2017) 1700090. DOI:10.1002/smtd.201700090 |
| [5] |
Y. Chen, X. Deng, J. Wen, J. Zhu, Z. Bian, Appl. Catal. B 258 (2019) 118024. DOI:10.1016/j.apcatb.2019.118024 |
| [6] |
J. Wen, L. Ling, Y. Chen, Z. Bian, Chin. J. Catal. 41 (2020) 1674-1681. DOI:10.1016/S1872-2067(20)63581-1 |
| [7] |
Y. Feng, M. Xu, H. Liu, et al., Nano Energy 73 (2020) 104768. DOI:10.1016/j.nanoen.2020.104768 |
| [8] |
X. Deng, Y. Chen, J. Wen, et al., Sci. Bull. (Beijing) 65 (2020) 105-112. DOI:10.1016/j.scib.2019.10.020 |
| [9] |
Y.F. Xu, H.S. Rao, B.X. Chen, et al., Adv. Sci. 2 (2015) 1500049. DOI:10.1002/advs.201500049 |
| [10] |
H. Tang, C.M. Hessel, J. Wang, et al., Chem. Soc. Rev. 43 (2014) 4281-4299. DOI:10.1039/C3CS60437C |
| [11] |
J. Luo, J.H. Im, M.T. Mayer, et al., Science 345 (2014) 1593-1596. DOI:10.1126/science.1258307 |
| [12] |
A. Le Goff, V. Artero, B. Jousselme, et al., Science 326 (2009) 1384-1387. DOI:10.1126/science.1179773 |
| [13] |
J.A. Turner, Science 305 (2004) 972-974. DOI:10.1126/science.1103197 |
| [14] |
T.R. Cook, D.K. Dogutan, S.Y. Reece, et al., Chem. Rev. 110 (2010) 6474-6502. DOI:10.1021/cr100246c |
| [15] |
M.G. Walter, E.L. Warren, J.R. McKone, et al., Chem. Rev. 110 (2010) 6446-6473. DOI:10.1021/cr1002326 |
| [16] |
S.T. Hunt, M. Milina, Z. Wang, Y. Román-Leshkov, Energy Environ. Sci. 9 (2016) 3290-3301. DOI:10.1039/C6EE01929C |
| [17] |
N.S. Lewis, Science 315 (2007) 798-801. DOI:10.1126/science.1137014 |
| [18] |
Y. Zheng, Y. Jiao, A. Vasileff, S.Z. Qiao, Angew. Chem. Int. Ed. 57 (2018) 7568-7579. DOI:10.1002/anie.201710556 |
| [19] |
S. Marini, P. Salvi, P. Nelli, et al., Electrochim. Acta 82 (2012) 384-391. DOI:10.1016/j.electacta.2012.05.011 |
| [20] |
K.F.L. Hagesteijn, S. Jiang, B.P. Ladewig, J. Mater, Sci. 53 (2018) 11131-11150. DOI:10.1007/s10853-018-2409-y |
| [21] |
I. Vincent, D. Bessarabov, Renewable Sustainable Energy Rev. 81 (2018) 1690-1704. DOI:10.1016/j.rser.2017.05.258 |
| [22] |
R. Subbaraman, D. Tripkovic, D. Strmcnik, et al., Science 334 (2011) 1256-1260. DOI:10.1126/science.1211934 |
| [23] |
Y. Shi, B. Zhang, Chem. Soc. Rev. 45 (2016) 1529-1541. DOI:10.1039/C5CS00434A |
| [24] |
J.W.D. Ng, M. García-Melchor, M. Bajdich, et al., Nat. Energy 1 (2016) 16053-16061. DOI:10.1038/nenergy.2016.53 |
| [25] |
B. Fei, Z. Chen, J. Liu, et al., Adv. Energy Mater. 10 (2020) 2001963. DOI:10.1002/aenm.202001963 |
| [26] |
D. Zhou, B. Jiang, R. Yang, X. Hou, C. Zheng, Chin. Chem. Lett. 31 (2020) 1540-1544. |
| [27] |
H. Zhou, F. Yu, Q. Zhu, et al., Energy Environ. Sci. 11 (2018) 2858-2864. DOI:10.1039/C8EE00927A |
| [28] |
L. Shao, H. Sun, L. Miao, et al., J. Mater. Chem. A: Mater. Energy Sustain. 6 (2018) 2494-2499. DOI:10.1039/C7TA10884B |
| [29] |
G. Wei, K. Du, X. Zhao, et al., Chin. Chem. Lett. 31 (2020) 2641-2644. DOI:10.1016/j.cclet.2020.02.029 |
| [30] |
J. Yu, T.A. Le, N.Q. Tran, H. Lee, Chem. 26 (2020) 6423-6436. DOI:10.1002/chem.202000209 |
| [31] |
J. Yin, J. Jin, H. Lin, et al., Adv. Sci. 7 (2020) 1903070. DOI:10.1002/advs.201903070 |
| [32] |
J. Zhu, L. Hu, P. Zhao, L.Y.S. Lee, K.Y. Wong, Chem. Rev. 120 (2019) 851-918. DOI:10.1021/acs.chemrev.9b00248 |
| [33] |
Q. Gao, W. Zhang, Z. Shi, L. Yang, Y. Tang, Adv. Mater. 31 (2019) 1802880. DOI:10.1002/adma.201802880 |
| [34] |
K. Wang, X. Wang, Z. Li, et al., Nano Energy 77 (2020) 105162. DOI:10.1016/j.nanoen.2020.105162 |
| [35] |
C. Lei, S. Lyu, J. Si, et al., ChemCatChem 11 (2019) 5855-5874. DOI:10.1002/cctc.201901707 |
| [36] |
J.Y. Wang, W.T. Liu, X.P. Li, T. Ouyang, Z.Q. Liu, Chem. Comm. 56 (2020) 1489-1492. DOI:10.1039/C9CC09303F |
| [37] |
J.Y. Wang, T. Ouyang, Y.P. Deng, Y.S. Hong, Z.Q. Liu, J. Power Sources 420 (2019) 108-117. DOI:10.1016/j.jpowsour.2019.02.098 |
| [38] |
L. Liao, X. Bian, J. Xiao, et al., Phys. Chem. Chem. Phys. 16 (2014) 10088-10094. DOI:10.1039/C3CP54754J |
| [39] |
R. Chen, S.F. Hung, D. Zhou, et al., Adv. Mater. 31 (2019) 1903909. DOI:10.1002/adma.201903909 |
| [40] |
X.P. Li, W.K. Han, K. Xiao, et al., Catal. Sci. Technol. 10 (2020) 4184-4190. DOI:10.1039/D0CY00315H |
| [41] |
L. Zeng, L. Yang, J. Lu, et al., Chin. Chem. Lett. 29 (2018) 1875-1878. DOI:10.1016/j.cclet.2018.10.026 |
| [42] |
B. Wang, C. Tang, H.F. Wang, et al., Small Methods 2 (2018) 1800055. DOI:10.1002/smtd.201800055 |
| [43] |
N.R. Chodankar, S.H. Ji, Y.K. Han, D.H. Kim, Nano-Micro Lett. 12 (2020) 1-12. DOI:10.1007/s40820-019-0337-2 |
| [44] |
X. Zhao, B. Pattengale, D. Fan, et al., ACS Energy Lett. 3 (2018) 2520-2526. DOI:10.1021/acsenergylett.8b01540 |
| [45] |
Y. Tong, H. Liu, M. Dai, L. Xiao, X. Wu, Chin. Chem. Lett. 31 (2020) 2295-2299. DOI:10.1016/j.cclet.2020.03.029 |
| [46] |
M. Hu, S. Zhao, S. Liu, et al., Adv. Mater. 30 (2018) 1801878. DOI:10.1002/adma.201801878 |
| [47] |
F.M. Zhang, J.L. Sheng, Z.D. Yang, et al., Angew. Chem. Int. Ed. 57 (2018) 12106-12110. DOI:10.1002/anie.201806862 |
| [48] |
D.H. Nam, O.S. Bushuyev, J. Li, et al., J. Am. Chem. Soc. 140 (2018) 11378-11386. DOI:10.1021/jacs.8b06407 |
| [49] |
J. Huang, Y. Sun, X. Du, et al., Adv. Mater. 30 (2018) 1803367. DOI:10.1002/adma.201803367 |
| [50] |
T. Ouyang, X.T. Wang, X.Q. Mai, et al., Angew. Chem. Int. Ed. 59 (2020) 11948-11957. DOI:10.1002/anie.202004533 |
| [51] |
H. Cheng, Y.Z. Su, P.Y. Kuang, G.F. Chen, Z.Q. Liu, J. Mater. Chem. A: Mater. Energy Sustain. 3 (2015) 19314-19321. DOI:10.1039/C5TA03985A |
| [52] |
H. Cheng, C.Y. Su, Z.Y. Tan, S.Z. Tai, Z.Q. Liu, J. Power Sources 357 (2017) 1-10. DOI:10.1016/j.jpowsour.2017.04.091 |
| [53] |
T. Ouyang, A.N. Chen, Z.Z. He, Z.Q. Liu, Y. Tong, Chem. Comm. 54 (2018) 9901-9904. DOI:10.1039/C8CC03975E |
| [54] |
Y. Zhou, S. Sun, J. Song, et al., Adv. Mater. 30 (2018) 1802912. DOI:10.1002/adma.201802912 |
| [55] |
Y. Yu, J. Zhou, Z. Sun, Adv. Funct. Mater. 30 (2020) 2000570. DOI:10.1002/adfm.202000570 |
| [56] |
S. Anantharaj, S.R. Ede, K. Sakthikumar, et al., ACS Catal. 6 (2016) 8069-8097. DOI:10.1021/acscatal.6b02479 |
| [57] |
S. Chandrasekaran, L. Yao, L. Deng, et al., Chem. Soc. Rev. 48 (2019) 4178-4280. DOI:10.1039/C8CS00664D |
| [58] |
D. He, X. Wu, W. Liu, et al., Chin. Chem. Lett. 30 (2019) 229-233. DOI:10.1016/j.cclet.2018.03.020 |
| [59] |
G.F. Chen, T.Y. Ma, Z.Q. Liu, et al., Adv. Funct. Mater. 26 (2016) 3314-3323. DOI:10.1002/adfm.201505626 |
| [60] |
M.R. Gao, J.X. Liang, Y.R. Zheng, et al., Nat. Commun. 6 (2015) 5982. DOI:10.1038/ncomms6982 |
| [61] |
X. Xu, F. Song, X. Hu, Nat. Commun. 7 (2016) 12324. DOI:10.1038/ncomms12324 |
| [62] |
J.Y. Wang, T. Ouyang, N. Li, T.Y. Ma, Z.Q. Liu, Sci. Bull. (Beijing) 63 (2018) 1130-1140. DOI:10.1016/j.scib.2018.07.008 |
| [63] |
L. Chai, Z. Hu, X. Wang, et al., Adv. Sci. 7 (2020) 1903195. DOI:10.1002/advs.201903195 |
| [64] |
L. Wu, L. Yu, F. Zhang, et al., Adv. Funct. Mater. (2020) 2006484. |
| [65] |
C. Huang, Y. Zou, Y.Q. Ye, et al., Chem. Comm. 55 (2019) 7687-7690. DOI:10.1039/C9CC03024G |
| [66] |
Y. Ji, J. Xie, Y. Yang, et al., Chin. Chem. Lett. 31 (2020) 855-858. DOI:10.1016/j.cclet.2019.06.021 |
| [67] |
F. Cheng, L. Wang, H. Wang, et al., Nano Energy 71 (2020) 104621. DOI:10.1016/j.nanoen.2020.104621 |
| [68] |
J.K. Nørskov, T. Bligaard, A. Logadottir, et al., J. Electrochem. Soc. 152 (2005) J23-J26. DOI:10.1149/1.1856988 |
| [69] |
J. Greeley, T.F. Jaramillo, J. Bonde, I.B. Chorkendorff, J.K. Norskov, Nat. Mater. 5 (2006) 909-913. DOI:10.1038/nmat1752 |
| [70] |
S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem. 39 (1972) 163-184. DOI:10.1016/S0022-0728(72)80485-6 |
| [71] |
J. Kang, J. Hwang, B. Han, J. Phys. Chem. C 122 (2018) 2107-2112. DOI:10.1021/acs.jpcc.7b09294 |
| [72] |
T.F. Jaramillo, K.P. Jorgensen, J. Bonde, et al., Science 317 (2007) 100-102. DOI:10.1126/science.1141483 |
| [73] |
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. DOI:10.1126/science.aad4998 |
| [74] |
D.R. Weinberg, C.J. Gagliardi, J.F. Hull, et al., Chem. Rev. 112 (2012) 4016-4093. DOI:10.1021/cr200177j |
| [75] |
W.T. Hong, M. Risch, K.A. Stoerzinger, et al., Energy Environ. Sci. 8 (2015) 1404-1427. DOI:10.1039/C4EE03869J |
| [76] |
M.T.M. Koper, J. Electroanal. Chem. (Lausanne) 660 (2011) 254-260. DOI:10.1016/j.jelechem.2010.10.004 |
| [77] |
M. Wohlfahrt-Mehrens, J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem. 237 (1987) 251-260. DOI:10.1016/0022-0728(87)85237-3 |
| [78] |
J.H. Montoya, L.C. Seitz, P. Chakthranont, et al., Nat. Mater. 16 (2016) 70-81. DOI:10.1038/nmat4778 |
| [79] |
S. Li, G. Zhang, X. Tu, J. Li, ChemElectroChem 5 (2018) 701-707. DOI:10.1002/celc.201701112 |
| [80] |
X. Ji, C. Cheng, Z. Zang, et al., J.Mater. Chem. A: Mater. Energy Sustain. 8 (2020) 21199-21207. DOI:10.1039/D0TA07676G |
| [81] |
H. Xu, S. Ci, Y. Ding, G. Wang, Z. Wen, J. Mater. Chem. A: Mater. Energy Sustain. 7 (2019) 8006-8029. DOI:10.1039/C9TA00833K |
| [82] |
H. Du, R.M. Kong, X. Guo, F. Qu, J. Li, Nanoscale 10 (2018) 21617-21624. DOI:10.1039/C8NR07891B |
| [83] |
K. Zeng, D. Zhang, Prog. Energy Combust. Sci. 36 (2010) 307-326. DOI:10.1016/j.pecs.2009.11.002 |
| [84] |
W. Wang, X. Xu, W. Zhou, Z. Shao, Adv. Sci. 4 (2017) 1600371. DOI:10.1002/advs.201600371 |
| [85] |
J. Rossmeisl, A. Logadottir, J.K. Nørskov, Chem. Phys. 319 (2005) 178-184. DOI:10.1016/j.chemphys.2005.05.038 |
| [86] |
J. Rossmeisl, Z.W. Qu, H. Zhu, G.J. Kroes, J.K. Nørskov, J. Electroanal. Chem. (Lausanne) 607 (2007) 83-89. DOI:10.1016/j.jelechem.2006.11.008 |
| [87] |
I.C. Man, H.Y. Su, F. Calle-Vallejo, et al., ChemCatChem 3 (2011) 1159-1165. DOI:10.1002/cctc.201000397 |
| [88] |
O. Diaz-Morales, I. Ledezma-Yanez, M.T.M. Koper, F. Calle-Vallejo, ACS Catal. 5 (2015) 5380-5387. DOI:10.1021/acscatal.5b01638 |
| [89] |
S. Trasatti, O.A. Petrii, J. Electroanal. Chem. (Lausanne) 327 (1992) 353-376. DOI:10.1016/0022-0728(92)80162-W |
| [90] |
H. Jin, C. Guo, X. Liu, et al., Chem. Rev. 118 (2018) 6337-6408. DOI:10.1021/acs.chemrev.7b00689 |
| [91] |
S. Czioska, J. Wang, X. Teng, Z. Chen, ACS Sustainable Chem. Eng. 6 (2018) 11877-11883. DOI:10.1021/acssuschemeng.8b02155 |
| [92] |
R. Karimi Shervedani, M. Torabi, F. Yaghoobi, Electrochim. Acta 244 (2017) 230-238. DOI:10.1016/j.electacta.2017.05.099 |
| [93] |
S. Wu, J. Liu, B. Cui, et al., Electrochim. Acta 299 (2019) 231-244. DOI:10.1016/j.electacta.2019.01.012 |
| [94] |
M. Sial, H. Lin, X. Wang, Nanoscale 10 (2018) 12975-12980. DOI:10.1039/C8NR03350A |
| [95] |
H. Cheng, M.L. Li, C.Y. Su, N. Li, Z.Q. Liu, Adv. Funct. Mater. 27 (2017) 1701833. DOI:10.1002/adfm.201701833 |
| [96] |
X.T. Wang, T. Ouyang, L. Wang, J.H. Zhong, Z.Q. Liu, Angew. Chem. Int. Ed. 59 (2020) 6492-6499. DOI:10.1002/anie.202000690 |
| [97] |
X.T. Wang, T. Ouyang, L. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 13291-13296. DOI:10.1002/anie.201907595 |
| [98] |
H. Su, X.T. Wang, X. J.-Hu, et al., J. Mater. Chem. A: Mater. Energy Sustain. 7 (2019) 22307-22313. DOI:10.1039/C9TA08064C |
| [99] |
X.X. Li, X.T. Wang, K. Xiao, et al., J. Power Sources 402 (2018) 116-123. DOI:10.1016/j.jpowsour.2018.09.021 |
| [100] |
X. Zou, J. Su, R. Silva, et al., Chem. Commun. (Camb.) 49 (2013) 7522-7524. DOI:10.1039/c3cc42891e |
| [101] |
J.A. Koza, Z. He, A.S. Miller, J.A. Switzer, Chem. Mater. 24 (2012) 3567-3573. DOI:10.1021/cm3012205 |
| [102] |
Y. Liang, Y. Li, H. Wang, et al., Nat. Mater. 10 (2011) 780-786. DOI:10.1038/nmat3087 |
| [103] |
Z. Chen, C.X. Kronawitter, B.E. Koel, Phys. Chem. Chem. Phys. 17 (2015) 29387-29393. DOI:10.1039/C5CP02876K |
| [104] |
H. Jin, J. Wang, D. Su, et al., J. Am. Chem. Soc. 137 (2015) 2688-2694. DOI:10.1021/ja5127165 |
| [105] |
G. Cheng, T. Kou, J. Zhang, et al., Nano Energy 38 (2017) 155-166. DOI:10.1016/j.nanoen.2017.05.043 |
| [106] |
L. Xu, Q. Jiang, Z. Xiao, et al., Angew. Chem. Int. Ed. 55 (2016) 5277-5281. DOI:10.1002/anie.201600687 |
| [107] |
T. Zhang, M.Y. Wu, D.Y. Yan, et al., Nano Energy 43 (2018) 103-109. DOI:10.1016/j.nanoen.2017.11.015 |
| [108] |
D. Yan, R. Chen, Z. Xiao, S. Wang, Electrochim. Acta 303 (2019) 316-322. DOI:10.1016/j.electacta.2019.02.091 |
| [109] |
Y. Jin, H. Wang, J. Li, et al., Adv. Mater. 28 (2016) 3785-3790. DOI:10.1002/adma.201506314 |
| [110] |
Y. Zhao, C. Chang, F. Teng, et al., Adv. Energy Mater. 7 (2017) 1700005. DOI:10.1002/aenm.201700005 |
| [111] |
C. Dong, T. Kou, H. Gao, Z. Peng, Z. Zhang, Adv. Energy Mater. 8 (2018) 1701347. DOI:10.1002/aenm.201701347 |
| [112] |
S. Peng, F. Gong, L. Li, et al., J. Am. Chem. Soc. 140 (2018) 13644-13653. DOI:10.1021/jacs.8b05134 |
| [113] |
J. Li, Y. Wang, T. Zhou, et al., J. Am. Chem. Soc. 137 (2015) 14305-14312. DOI:10.1021/jacs.5b07756 |
| [114] |
W.F. Chen, C.H. Wang, K. Sasaki, et al., Energy Environ. Sci. 6 (2013) 943-951. DOI:10.1039/c2ee23891h |
| [115] |
L. Liao, S. Wang, J. Xiao, et al., Energy Environ. Sci. 7 (2014) 387-392. DOI:10.1039/C3EE42441C |
| [116] |
D.V. Esposito, S.T. Hunt, Y.C. Kimmel, J.G. Chen, J. Am. Chem. Soc. 134 (2012) 3025-3033. DOI:10.1021/ja208656v |
| [117] |
K. Xiong, L. Li, L. Zhang, et al., J. Mater. Chem. A: Mater. Energy Sustain. 3 (2015) 1863-1867. DOI:10.1039/C4TA05686H |
| [118] |
R. Ma, Y. Zhou, Y. Chen, et al., Angew. Chem. Int. Ed. 54 (2015) 14723-14727. DOI:10.1002/anie.201506727 |
| [119] |
H. Wang, C. Sun, Y. Cao, et al., Carbon 114 (2017) 628-634. DOI:10.1016/j.carbon.2016.12.081 |
| [120] |
H. Lin, Z. Shi, S. He, et al., Chem. Sci. 7 (2016) 3399-3405. DOI:10.1039/C6SC00077K |
| [121] |
Y. Zhao, K. Kamiya, K. Hashimoto, S. Nakanishi, J. Am. Chem. Soc. 137 (2014) 110-113. DOI:10.1021/ja5114529 |
| [122] |
J.S. Li, Y. Wang, C.H. Liu, et al., Nat. Commun. 7 (2016) 11204. DOI:10.1038/ncomms11204 |
| [123] |
H.B. Wu, B.Y. Xia, L. Yu, X.Y. Yu, X.W. Lou, Nat. Commun. 6 (2015) 6512. DOI:10.1038/ncomms7512 |
| [124] |
H. Vrubel, X. Hu, Angew. Chem. Int. Ed. 51 (2012) 12703-12706. DOI:10.1002/anie.201207111 |
| [125] |
F.X. Ma, H.B. Wu, B.Y. Xia, C.Y. Xu, X.W. Lou, Angew. Chem. Int. Ed. 54 (2015) 15395-15399. DOI:10.1002/anie.201508715 |
| [126] |
J.T. Ren, L. Chen, C.C. Weng, G.G. Yuan, Z.Y. Yuan, ACS Appl. Mater. Interfaces 10 (2018) 33276-33286. DOI:10.1021/acsami.8b12108 |
| [127] |
Z. Kou, L. Zhang, Y. Ma, et al., Appl. Catal. B 243 (2019) 678-685. DOI:10.1016/j.apcatb.2018.11.008 |
| [128] |
H. Wang, Y. Cao, C. Sun, et al., ChemSusChem. 10 (2017) 3540-3546. DOI:10.1002/cssc.201701276 |
| [129] |
J.R. McKone, B.F. Sadtler, C.A. Werlang, N.S. Lewis, H.B. Gray, ACS Catal. 3 (2013) 166-169. DOI:10.1021/cs300691m |
| [130] |
W.F. Chen, J.T. Muckerman, E. Fujita, Chem. Commun. (Camb.) 49 (2013) 8896-8909. DOI:10.1039/c3cc44076a |
| [131] |
Y. Yang, J. Liu, S. Guo, Y. Liu, Z. Kang, J.Mater. Chem. A: Mater.Energy Sustain. 3 (2015) 18598-18604. DOI:10.1039/C5TA04867B |
| [132] |
M. Gong, W. Zhou, M.C. Tsai, et al., Nat. Commun. 5 (2014) 4695. DOI:10.1038/ncomms5695 |
| [133] |
R. Subbaraman, D. Tripkovic, K.C. Chang, et al., Nat. Mater. 11 (2012) 550-557. DOI:10.1038/nmat3313 |
| [134] |
Z.Y. Yu, Y. Duan, M.R. Gao, et al., Chem. Sci. 8 (2017) 968-973. DOI:10.1039/C6SC03356C |
| [135] |
M. Li, Y. Zhu, H. Wang, et al., Adv. Energy Mater. 9 (2019) 1803185. DOI:10.1002/aenm.201803185 |
| [136] |
Y. Zhao, K. Kamiya, K. Hashimoto, S. Nakanishi, Angew. Chem. Int. Ed. 52 (2013) 13638-13641. DOI:10.1002/anie.201307527 |
| [137] |
W.F. Chen, J.M. Schneider, K. Sasaki, et al., ChemSusChem. 7 (2014) 2414-2418. DOI:10.1002/cssc.201402454 |
| [138] |
N. Han, K.R. Yang, Z. Lu, et al., Nat. Commun. 9 (2018) 924. DOI:10.1038/s41467-018-03429-z |
| [139] |
Z.J. Chen, G.X. Cao, L.Y. Gan, et al., ACS Catal. 8 (2018) 8866-8872. DOI:10.1021/acscatal.8b02212 |
| [140] |
J. Zhang, Y. Liu, C. Sun, et al., ACS Energy Lett. 3 (2018) 779-786. DOI:10.1021/acsenergylett.8b00066 |
| [141] |
X. Xiong, C. You, Z. Liu, A.M. Asiri, X. Sun, ACS Sustainable Chem. Eng. 6 (2018) 2883-2887. DOI:10.1021/acssuschemeng.7b03752 |
| [142] |
N. Jiang, B. You, M. Sheng, Y. Sun, Angew. Chem. Int. Ed. 54 (2015) 6251-6254. DOI:10.1002/anie.201501616 |
| [143] |
T.H.M. Lau, X. Lu, J. Kulhavy, et al., Chem. Sci. 9 (2018) 4769-4776. DOI:10.1039/C8SC01114A |
| [144] |
L. Xie, F. Qu, Z. Liu, et al., J. Mater. Chem. A: Mater. Energy Sustain. 5 (2017) 7806-7810. DOI:10.1039/C7TA02333B |
| [145] |
J. Chen, B. Ren, H. Cui, C. Wang, Small 16 (2020) 1907556. DOI:10.1002/smll.201907556 |
| [146] |
S. Zhang, G. Gao, J. Hao, et al., ACS Appl. Mater. Interfaces 11 (2019) 43261-43269. DOI:10.1021/acsami.9b16390 |
| [147] |
P. Liu, J.A. Rodriguez, J. Am. Chem. Soc. 127 (2005) 14871-14878. DOI:10.1021/ja0540019 |
| [148] |
A.E. Henkes, Y. Vasquez, R.E. Schaak, J. Am. Chem. Soc. 129 (2007) 1896-1897. DOI:10.1021/ja068502l |
| [149] |
E.J. Popczun, C.G. Read, C.W. Roske, N.S. Lewis, R.E. Schaak, Angew. Chem. Int. Ed. 53 (2014) 5427-5430. DOI:10.1002/anie.201402646 |
| [150] |
P. Liu, J.A. Rodriguez, T. Asakura, J. Gomes, K. Nakamura, J. Phys. Chem. B 109 (2005) 4575-4583. |
| [151] |
G. Zhang, G. Wang, Y. Liu, et al., J. Am. Chem. Soc. 138 (2016) 14686-14693. DOI:10.1021/jacs.6b08491 |
| [152] |
J. Kibsgaard, T.F. Jaramillo, Angew. Chem. Int. Ed. 53 (2014) 14433-14437. DOI:10.1002/anie.201408222 |
| [153] |
J. Kibsgaard, C. Tsai, K. Chan, et al., Energy Environ. Sci. 8 (2015) 3022-3029. DOI:10.1039/C5EE02179K |
| [154] |
E.J. Popczun, J.R. McKone, C.G. Read, et al., J. Am. Chem. Soc. 135 (2013) 9267-9270. DOI:10.1021/ja403440e |
| [155] |
Z. Huang, Z. Chen, Z. Chen, et al., Nano Energy 9 (2014) 373-382. DOI:10.1016/j.nanoen.2014.08.013 |
| [156] |
D.H. Ha, B. Han, M. Risch, et al., Nano Energy 29 (2016) 37-45. DOI:10.1016/j.nanoen.2016.04.034 |
| [157] |
Y.Y. Cai, X.H. Li, Y.N. Zhang, et al., Angew. Chem. Int. Ed. 52 (2013) 11822-11825. DOI:10.1002/anie.201304652 |
| [158] |
X.H. Li, M. Antonietti, Chem. Soc. Rev. 42 (2013) 6593-6604. DOI:10.1039/c3cs60067j |
| [159] |
X.H. Li, Y.Y. Cai, L.H. Gong, et al., Chem. 20 (2014) 16732-16737. DOI:10.1002/chem.201404325 |
| [160] |
L.B. Lv, T.N. Ye, L.H. Gong, et al., Chem. Mater. 27 (2015) 544-549. DOI:10.1021/cm503988n |
| [161] |
Z.H. Xue, H. Su, Q.Y. Yu, et al., Adv. Energy Mater. 7 (2017) 1602355. DOI:10.1002/aenm.201602355 |
| [162] |
R. Wu, B. Xiao, Q. Gao, et al., Angew. Chem. Int. Ed. 57 (2018) 15445-15449. DOI:10.1002/anie.201808929 |
| [163] |
C. Huang, T. Ouyang, Y. Zou, N. Li, Z.Q. Liu, J. Mater. Chem. A: Mater. Energy Sustain. 6 (2018) 7420-7427. DOI:10.1039/C7TA11364A |
| [164] |
Y. Li, H. Zhang, M. Jiang, et al., Adv. Funct. Mater. 27 (2017) 1702513. DOI:10.1002/adfm.201702513 |
| [165] |
Y. Wu, X. Tao, Y. Qing, et al., Adv. Mater. 31 (2019) 1900178. DOI:10.1002/adma.201900178 |
| [166] |
L.M. Cao, Y.W. Hu, S.F. Tang, et al., Adv. Sci. 5 (2018) 1800949. DOI:10.1002/advs.201800949 |
| [167] |
B. Zhang, Y.H. Lui, H. Ni, S. Hu, Nano Energy 38 (2017) 553-560. DOI:10.1016/j.nanoen.2017.06.032 |
| [168] |
L. Yan, B. Zhang, J. Zhu, et al., Appl. Catal. B 265 (2020) 118555. DOI:10.1016/j.apcatb.2019.118555 |
| [169] |
W. Zhu, X. Yue, W. Zhang, et al., Chem. Commun. (Camb.) 52 (2016) 1486-1489. DOI:10.1039/C5CC08064A |
| [170] |
C. Ouyang, X. Wang, C. Wang, et al., Electrochim. Acta 174 (2015) 297-301. DOI:10.1016/j.electacta.2015.05.186 |
| [171] |
B. Chen, R. Li, G. Ma, et al., Nanoscale 7 (2015) 20674-20684. DOI:10.1039/C5NR07429K |
| [172] |
J.M. Falkowski, N.M. Concannon, B. Yan, Y. Surendranath, J. Am. Chem. Soc. 137 (2015) 7978-7981. DOI:10.1021/jacs.5b03426 |
| [173] |
R. Wu, J. Zhang, Y. Shi, D. Liu, B. Zhang, J. Am. Chem. Soc. 137 (2015) 6983-6986. DOI:10.1021/jacs.5b01330 |
| [174] |
K. Xu, P. Chen, X. Li, et al., J. Am. Chem. Soc. 137 (2015) 4119-4125. DOI:10.1021/ja5119495 |
| [175] |
N. Jiang, L. Bogoev, M. Popova, et al., J. Mater. Chem. A: Mater. Energy Sustain. 2 (2014) 19407-19414. DOI:10.1039/C4TA04339A |
| [176] |
W. Zhou, X.J. Wu, X. Cao, et al., Energy Environ. Sci. 6 (2013) 2921-2924. DOI:10.1039/c3ee41572d |
| [177] |
P. Chen, T. Zhou, M. Zhang, et al., Adv. Mater. 29 (2017) 1701584. DOI:10.1002/adma.201701584 |
| [178] |
Z. Quan, Y. Wang, J. Fang, Acc. Chem. Res. 46 (2013) 191-202. DOI:10.1021/ar200293n |
| [179] |
G. Liu, H.G. Yang, J. Pan, et al., Chem. Rev. 114 (2014) 9559-9612. DOI:10.1021/cr400621z |
| [180] |
L.L. Feng, G. Yu, Y. Wu, et al., J. Am. Chem. Soc. 137 (2015) 14023-14026. DOI:10.1021/jacs.5b08186 |
| [181] |
C. Tsai, F. Abild-Pedersen, J.K. Norskov, Nano Lett. 14 (2014) 1381-1387. DOI:10.1021/nl404444k |
| [182] |
D. Voiry, R. Fullon, J. Yang, et al., Nat. Mater. 15 (2016) 1003-1009. DOI:10.1038/nmat4660 |
| [183] |
M.A. Lukowski, A.S. Daniel, F. Meng, et al., J. Am. Chem. Soc. 135 (2013) 10274-10277. DOI:10.1021/ja404523s |
| [184] |
Q. Liu, X. Li, Q. He, et al., Small 11 (2015) 5556-5564. DOI:10.1002/smll.201501822 |
| [185] |
Q. Xiong, Y. Wang, P.F. Liu, et al., Adv. Mater. 30 (2018) 1801450. DOI:10.1002/adma.201801450 |
| [186] |
Y. Huang, Y. Sun, X. Zheng, et al., Nat. Commun. 10 (2019) 982. DOI:10.1038/s41467-019-08877-9 |
| [187] |
S. Huang, Y. Meng, S. He, et al., Adv. Funct. Mater. 27 (2017) 1606585. DOI:10.1002/adfm.201606585 |
| [188] |
S. Gupta, S. Zhao, X.X. Wang, et al., ACS Catal. 7 (2017) 8386-8393. DOI:10.1021/acscatal.7b02949 |
| [189] |
M. Kuang, Q. Wang, H. Ge, et al., ACS Energy Lett. 2 (2017) 2498-2505. DOI:10.1021/acsenergylett.7b00835 |
| [190] |
B. Zhang, X. Zheng, O. Voznyy, et al., Science 352 (2016) 333-337. DOI:10.1126/science.aaf1525 |
| [191] |
J. Zhang, L. Yu, Y. Chen, et al., Adv. Mater. 32 (2020) 1906432. DOI:10.1002/adma.201906432 |
| [192] |
J. Deng, Y. Su, D. Liu, et al., Chem. Rev. 119 (2019) 9221-9259. DOI:10.1021/acs.chemrev.9b00232 |
| [193] |
J. Wang, H. Cheng, S. Ren, et al., J. Mater. Chem. A: Mater. Energy Sustain. 8 (2020) 16018-16023. DOI:10.1039/D0TA03736B |
| [194] |
H. Yang, J. Bright, S. Kasani, et al., Nano Res. 12 (2018) 643-650. |
| [195] |
B. Lv, J. Jiao, Y. Liu, et al., Nanoscale 11 (2019) 22730-22733. DOI:10.1039/C9NR08571H |
| [196] |
A. Bergmann, T.E. Jones, E. Martinez Moreno, et al., Nat. Catal. 1 (2018) 711-719. DOI:10.1038/s41929-018-0141-2 |
| [197] |
J. Jiao, W. Yang, Y. Pan, et al., Small 16 (2020) 2002124. DOI:10.1002/smll.202002124 |
| [198] |
D.Y. Chung, J.W. Han, D.H. Lim, et al., Nanoscale 7 (2015) 5157-5163. DOI:10.1039/C4NR07648F |
| [199] |
Y. Wu, G.D. Li, Y. Liu, et al., Adv. Funct. Mater. 26 (2016) 4839-4847. DOI:10.1002/adfm.201601315 |
| [200] |
S. Deng, Y. Zhong, Y. Zeng, et al., Adv. Sci. 5 (2018) 1700772. DOI:10.1002/advs.201700772 |
| [201] |
F. Yu, H. Zhou, Y. Huang, et al., Nat. Commun. 9 (2018) 2551. DOI:10.1038/s41467-018-04746-z |
| [202] |
L. An, J. Feng, Y. Zhang, et al., Adv. Funct. Mater. 29 (2019) 1805298. DOI:10.1002/adfm.201805298 |
| [203] |
Y. Liu, S. Jiang, S. Li, et al., Appl. Catal. B 247 (2019) 107-114. DOI:10.1016/j.apcatb.2019.01.094 |
| [204] |
J. Zhang, T. Wang, D. Pohl, et al., Angew. Chem. Int. Ed. 55 (2016) 6702-6707. DOI:10.1002/anie.201602237 |
| [205] |
Y. Yang, K. Zhang, H. Lin, et al., ACS Catal. 7 (2017) 2357-2366. DOI:10.1021/acscatal.6b03192 |
| [206] |
Y. Guo, J. Tang, Z. Wang, et al., Nano Energy 47 (2018) 494-502. DOI:10.1016/j.nanoen.2018.03.012 |
| [207] |
Y. Yang, H. Yao, Z. Yu, et al., J. Am. Chem. Soc. 141 (2019) 10417-10430. DOI:10.1021/jacs.9b04492 |
| [208] |
Z. Kou, T. Wang, Q. Gu, et al., Adv. Energy Mater. 9 (2019) 1803768. DOI:10.1002/aenm.201803768 |
| [209] |
Q. Liang, H. Jin, Z. Wang, et al., Nano Energy 57 (2019) 746-752. DOI:10.1016/j.nanoen.2018.12.060 |
| [210] |
J. Lin, P. Wang, H. Wang, et al., Adv. Sci. 6 (2019) 1900246. DOI:10.1002/advs.201900246 |
| [211] |
L. Yang, R. Liu, L. Jiao, Adv. Funct. Mater. 30 (2020) 1909618. DOI:10.1002/adfm.201909618 |
| [212] |
C. Wu, B. Liu, J. Wang, et al., Appl. Surf. Sci. 441 (2018) 1024-1033. DOI:10.1016/j.apsusc.2018.02.076 |
| [213] |
X. Wang, W. Zhang, J. Zhang, Z. Wu, ChemElectroChem 6 (2019) 4550-4559. DOI:10.1002/celc.201901201 |
| [214] |
X. Luo, P. Ji, P. Wang, et al., Adv. Energy Mater. 10 (2020) 1903891. DOI:10.1002/aenm.201903891 |