 2021, Vol. 32
2021, Vol. 32 


Quinazoline is a preeminent class of structural motif in natural products, functional materials and bioactive compounds [1]. Among this family, 4-aminoquinazolines have drawn considerable attention due to their extensive occurrence in many medicinal molecules, such as inhibitors of breast cancer resistance protein (ABCG2) [2], antihypertensive drugs [3], anti-cancer agents [4] and importantly lapatinib [5] (Fig. 1). Because of their great value, the synthesis of 4-aminoquinazolines has gained much attention [6]. The traditional approach to 4-aminoquinazolines includes nucleophilic substitution of aryl or alkyl amines with 4-chloroquinazolines, which requires multi steps from 2-aminobenzoic acids [7]. Recently, Zhu's group developed a palladium-catalyzed intramolecular C(sp2)-H amidation to construct 4-aminoquinazolines in the presence of O2 as the terminal oxidant and starting from isonitriles [8]. Consequently, the development of greener pathway starting from readily available and easily handled reactants to access such a motif is of great significance.

|
Download:
|
| Fig. 1. Examples of 4-aminoquinazolines with bioactivity. | |
Transition-metal catalyzed annulation based on C-H activation has been considered as an efficient and atom-economic strategy for the construction of heterocyclic compounds in past decade [9]. Directing groups (DGs) are generally required to resolve the regio-selectivity and improve the catalytic activity of metal. To address the limitation of the DGs, such as requirement of preinstallation and removal, multiple functions has been imparted to DGs so that they could function as a nucleophilic or electrophilic reagent for the postchemical transformations as well as being a chelating group and internal oxidant [10]. Based on that, the commercially available imidamides have attracted intensive interest as substrates, because N-H imine could act as both directing groups and intramolecular nucleophile or electrophile [11]. In which, C-H regioselectivity presented challenge, however afforded the opportunity to manipulate an array of different heterocycles. Li, Wu and our group have developed Rh, Ir and Ru-catalyzed intramolecular annulations using N-H imine as directing group to construct indoles [12] and benzimidazoles [13]. In these reactions, the C-alkyl group promoted the selectivity and the metal-X (X = C, N) underwent migratory insertion into the C=N bond (acting an electrophile), causing NH to be removed through subsequent elimination. Imidamide was employed as four-atom synthon in the field of C-H activation, mainly occurred at N-phenyl ring instead of the C-phenyl ring perhaps due to its low thermos-stability of the key intermediate. So far, there are few successful reports from Li, Du and Liu groups on the Rh(Ⅲ)-catalyzed intermolecular C-H functionalization of N-phenylbenzimidamides with alkynes, diazo compounds and sulfur ylides to produce 1-aminoisoquinolines [14]. In these reactions, the Rh preferred to afford a five-membered metallacyclo-intermediate and the C-H activation selectively occurred at C-phenyl ring. With our ongoing efforts to build structurally divers heterocycles [15], herein, we explored a Rh(III)-catalyzed [4 + 2] cyclization of N-phenylbenzimidamides with 1, 4, 2-dioxazol-5-ones to synthesize 4-aminoquinazolines. C-H activation occurred at C-phenyl ring with excellent regio-selectivity. In this reaction, various 4-aminoquinazoline derivatives could be easily obtained with high step economy, and the title products could undergo further chemical transformation for the synthesis of useful nitrogen-heterocycles.
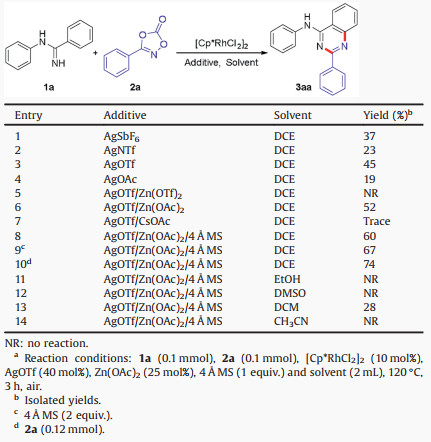
We initiated our study by using N-phenylbenzamidine 1a and 3-phenyl-1, 4, 2-dioxazol-5-one 2a as the model substrates to examine the reaction parameters. The desired [4 + 2] annulation occurred at C-phenyl ring to afford the product 3aa in 37% yield in the presence of [Cp*RhCl2]2 and AgSbF6 in DCE (1, 2-dichloroethane) at 120 ℃ under air for 3h (Table 1, entry 1). The structure and regioselectivity was unambiguously confirmed by the single-crystal X-ray diffraction analysis of 3aa (Supporting information). Screening the silver salts revealed that AgOTf was the best choice, and produced the desired product in 45% yield (Table 1, entries 2–4). Subsequently, a variety of additives were investigated and Zn(OAc)2 gave better results (Table 1, entries 5–7). After introduction of 4ÅMS as an additive, the yield of product reached to 60% (Table 1, entry 8). When the amount of 4ÅMS was increased to 2 equiv., 3aa could be afforded in 67% yield (Table 1, entry 9). Meanwhile, the higher yield was obtained by increasing the ratio of 1a and 2a to 1:1.2 (Table 1, entry 10). When the reactions were carried out in other medium, including EtOH (ethanol), DMSO (dimethyl sulfoxide), DCM (dichloromethane) and CH3CN (acetonitrile), the yield of the desired product was significantly decreased (Table 1, entries 11–14). Finally, the optimal reaction conditions were identified as follows: N-phenylbenzamidine (0.1 mmol), 3-phenyl-1, 4, 2-dioxazol-5-one(0.12 mmol), [Cp*RhCl2]2 (10 mol%), AgOTf (40 mol%), Zn(OAc)2 (25 mol%) and 4 Å MS (2 equiv.) in DCE (2 mL) at 120 ℃ under air atmosphere for 3 h.
|
|
Table 1 Optimization of the reaction conditions.a |
{kind=link}
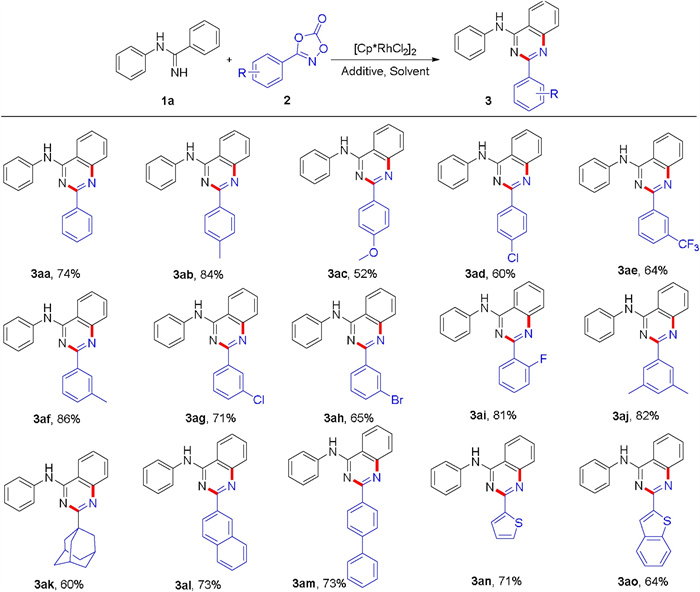
With the optimized conditions in hand, the substituted N-arylbenzamidines were first examined with 3-phenyl-1, 4, 2-dioxazol-5-one 2a (Scheme 1). To our delight, electron-donating (Me, OMe, Et, iPr, tBu) and electron-withdrawing groups (F, Cl, Br) at the para-position of N-phenyl ring worked well in this transformation, affording the desired products in 62%–75% yields (3aa-3ia). However, only trace amount of the desired product was observed for N-(4-NO2-phenyl)benzimidamide. The meta- (Me, OMe, F and Cl) and ortho-substituted N-arylbenzamidines (Me, F) were also compatible in this transformation, affording the corresponding products in 53%–78% yields (3ja-3oa). Meanwhile, the disubstituted N-arylbenzamidines were found to be amenable substrates, providing the products in 74%–76% yields (3pa-3qa). These results indicated that the steric hindrance of N-phenyl ring did not dramatically influence this transformation. N-Pyridyl and N-phenyl(4-benzyl) benzimidamides were all converted into the desired products in moderate yields (3ra-3sa). The N-alkylbenzamidine was investigated, such as n-propyl. However, no desired product was obtained. Moreover, substituents at the para and meta-position of C-phenyl ring also performed well, affording the product in 62%–78% yields (3ta-3wa). However, only trace amounts of desired products were obtained for ortho-Cl and ortho-Me substituted N-arylbenzamidines (3xa and 3ya), perhaps due to the steric hindrance.

|
Download:
|
| Scheme 1. Scope of the substituted N-arylbenzamidines. Reaction conditions: 1 (0.1 mmol), 2a (0.12 mmol), [Cp*RhCl2]2 (10 mol%), AgOTf (40 mol%), Zn(OAc)2 (25 mol%), 4 Å MS (2 equiv.) and DCE (2 mL), 120 ℃, 3 h, air. Isolated yields. | |
{kind=link}
To further explore the scope of this transformation, various 1, 4, 2-dioxazol-5-ones were investigated under the optimized conditions (Scheme 2). The various para- and meta-substituted (including Me, OMe, Cl, Br and CF3) 1, 4, 2-dioxazol-5-ones could couple successfully with N-phenylbenzamidine to deliver the corresponding products in 52%–86% yields (3aa-3ah). Meanwhile, the ortho- and poly-substituted 1, 4, 2-dioxazol-5-ones also performed well, affording the products in 81%–92% yields (3ai-3aj). These results indicated that steric hindrance of 1, 4, 2-dioxazol-5-ones did not dramatically influence this transformation. Satisfyingly, the reaction also displayed an excellent tolerance toward the group of adamantine (3ak). However, this catalytic system was not applied to the 3-pentyl-1, 4, 2-dioxazol-5-one. When the substrates with groups of naphthalene, bisphenyl, thiophene and benzothiophene, the transformation showed excellent tolerance and afforded 64%–73% yields (3al-3ao).

|
Download:
|
| Scheme 2. Scope of 1, 4, 2-dioxazol-5-ones. Reaction conditions: 1a (0.1 mmol), 2 (0.12 mmol), [Cp*RhCl2]2 (10 mol%), AgOTf (40 mol%), Zn(OAc)2 (25 mol%), 4 Å MS (2 equiv.) and DCE (2 mL), 120 ℃, 3 h, air. Isolated yields. | |
{kind=link}
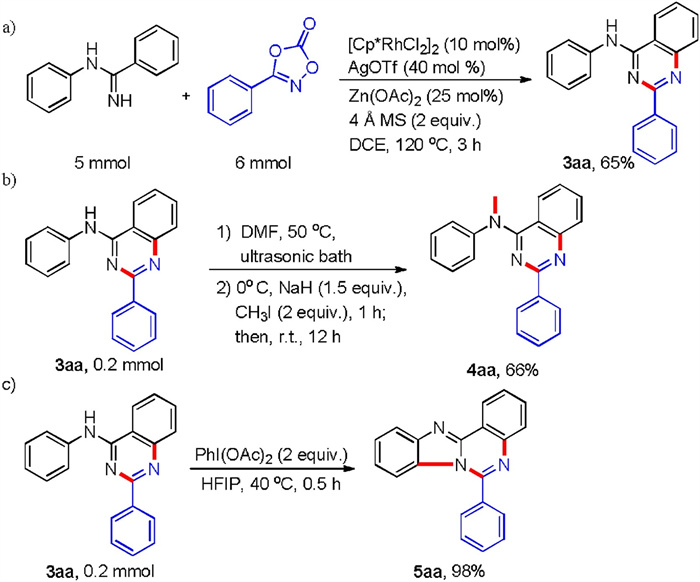
In addition, to demonstrate the synthetic utility of this transformation in organic synthesis, the template reaction was conducted with larger scale under the standard reaction conditions, and afforded 65% of isolated yield, confirming the robustness of these conditions (Scheme 3a). Meanwhile, we investigated protection of NH group in 3aa via coupling with iodo methane (Scheme 3b). The coupled product can be used as ligands in polymer light-emitting diodes [16]. Notably, the benzimidazo[1, 2-c]quinazolines 5aa could also be synthesized via a phenyliodine diacetate (PIDA)-mediated intramolecular C-H cycloamination of 3aa (Scheme 3c), and its derivatives exhibited excellent antibacterial and antifungal activities [17]. These results suggested that our strategy was a practicability synthetic method and a late-stage modification tool.

|
Download:
|
| Scheme 3. Scale-up reaction and derivation. | |
{kind=link}
Subsequently, a series of control experiments were conducted in order to further probe the reaction mechanism (Scheme 4). The hydrogen-deuterium exchange experiment of 1a was investigated in DCE/CD3OD (v/v = 9:1) under standard conditions for 10 min and the C-phenyl ring was observed with 60% D (Scheme 4a). It indicated that C-H activation might be a reversible progress and preferred to occur at the C-phenyl ring under the Rh(III) catalytic system, which is consistent with the results obtained. Meanwhile, the kinetic isotope effect (KIE=3.9) was obtained (Scheme 4b), indicating that the C-H activation might be involved in the turnover-limiting step.

|
Download:
|
| Scheme 4. Mechanistic studies. | |
{kind=link}
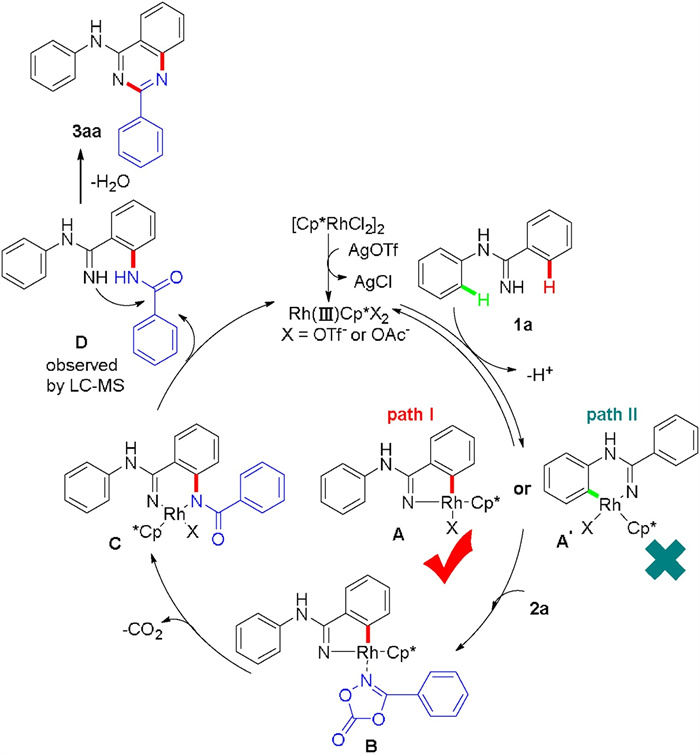
On the basis of above mechanistic investigations and the previous reported literatures [11k, 14d], a plausible mechanism of the [4 + 2] cycloaddition reaction is proposed in Scheme 5. Initially, the ligand exchange of [Cp*RhCl2]2 in the presence of OTf− and OAc- yielded active [Cp*RhX2] (X=OTf-, OAc-) species. Subsequently, a reversible C-H bond cleavage with N-phenylbenzamidine 1a afforded a five-membered cyclo-rhodium intermediate A instead of A′. Next, coordination of 3-phenyl-1, 4, 2-dioazol-5-one 2a with intermediate A gave the intermediate B, which underwent migratory insertion to generate the intermediate C by the elimination of CO2. Finally, protonation of C regenerated active complex of [Cp*RhX2] for the next catalytic cycle, and simultaneously resulted in the amidated product D, which underwent intramolecular nucleophile addition to give the desired product 3aa.

|
Download:
|
| Scheme 5. Proposed reaction mechanism. | |
{kind=link}
In summary, we have developed a novel and highly efficient Rhodium(III)-catalyzed [4 + 2] cyclization of N-arylbenzamidines and 1, 4, 2-dioxazol-5-ones to synthesize 4-aminoquinazolines via highly selective C-H bond functionalization at C-phenyl ring. This strategy proceeded with excellent regioselectivity, broad substrate scope and high step economy. Moreover, two C-N bonds were formed in reaction. The products were also subjected to various chemical transformations to afford the medicinal molecules, and may provide wider application in organic synthesis. Further research on the application of the 4-aminoquinazoline frameworks is currently underway.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe greatly acknowledge partial financial support from the Ministry of Science and Technology of China (No. 2016YFE0132600), Henan Center for Outstanding Overseas Scientists (No. GZS2020001), and Zhengzhou University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/jcclet.2021.02.061.
| [1] |
(a) J.P. Michael, Nat. Prod. Rep. 25 (2008) 166–187; (b) R. Gundla, R. Kazemi, R. Sanam, et al., J. Med. Chem. 51 (2008) 3367–3377; (c) M.J. Hour, J.S. Yang, T.L. Chen, et al., Eur. J. Med. Chem. 46 (2011) 2709–2721; (d) M.N.N. Lima, G.C. Cassiano, K.C.P. Tomaz, et al., Front. Chem. 7 (2019) 773; (e) R.L. Clements, V. Streva, P. Dumoulin, et al., J. Infect. Dis. 221 (2020) 956–962; (f) F. Wu, L. Zhuo, F. Wang, et al., iScience 23 (2020) 101179. |
| [2] |
(a) K. Juvale, J. Gallus, M. Wiese, Bioorg. Med. Chem. 21 (2013) 7858–7873; (b) M.K. Krapf, M. Wiese, J. Med. Chem. 59 (2016) 5449–5461; (c) M.K. Krapf, J. Gallus, M. Wiese, J. Med. Chem. 60 (2017) 4474–4495; (d) M.K. Krapf, J. Gallus, V. Namasivayam, M. Wiese, J. Med. Chem. 61 (2018) 7952–7976; (e) M.K. Krapf, J. Gallus, A. Spindler, M. Wiese, J. Med. Chem. 161 (2019) 506–525. |
| [3] |
(a) S.F. Campbell, M.J. Davey, J.D. Hardstone, B.N. Lewis, M.J. Palmer, J. Med. Chem. 30 (1987) 49–57; (b) S. Paliwal, A. Mittal, M. Sharma, et al., Med. Chem. Res. 24 (2015) 576–587. |
| [4] |
M.J. Hour, J.S. Yang, T.L. Chen, et al., Eur. J. Med. Chem. 46 (2011) 2709-2721. DOI:10.1016/j.ejmech.2011.03.059 |
| [5] |
B. Moy, P. Kirkpatrick, S. Kar, P. Goss, Nat. Rev. Drug Discov. 6 (2007) 431-432. DOI:10.1038/nrd2332 |
| [6] |
(a) F.C. Jia, Z.W. Zhou, C. Xu, et al., Org. Lett. 17 (2015) 4236–4239; (b) V. Este'vez, G.V. Baelen, B.H. Lentferink, et al., ACS Catal. 4 (2014) 40–43; (c) D.S. Yoon, Y. Han, T.M. Stark, et al., Org. Lett. 6 (2004) 4775–4778; (d) X.Y. Mao, X.T. Lin, M. Yang, G.S. Chen, Y.L. Lin, Adv. Synth. Catal. 360 (2018) 3643–3648. |
| [7] |
(a) N.Y. Kim, C.H. Cheon, Tetrahedron Lett. 55 (2014) 2340–2344; (b) M. Staderini, M.L. Bolognesi, J.C. Men[75_TD$DIF]éndez, Adv. Syn. Catal. 357 (2015) 185–195. |
| [8] |
Y. Wang, H. Wang, J. Peng, Q. Zhu, Org. Lett. 13 (2011) 4604-4607. DOI:10.1002/chin.201201140 |
| [9] |
(a) W. Yang, S. Ye, D. Fanning, et al., Angew. Chem. Int. Ed. 54 (2015) 2501–2504; (b) X. Wang, Y. Li, T. Knecht, et al., Angew. Chem. Int. Ed. 57 (2018) 5520–5524; (c) Y. Hwang, Y. Park, Y.B. Kim, D. Kim, S. Chang, Angew. Chem. Int. Ed. 57 (2018) 13565–13569; (d) R. Mi, G. Zheng, Z. Qi, X. Li, Angew. Chem. Int. Ed. 58 (2019) 17666–17670; (e) G. Zheng, J. Sun, Y. Xu, S. Zhai, X. Li, Angew. Chem. Int. Ed. 58 (2019) 5090–5094; (f) J.S. Ham, B. Park, M. Son, et al., J. Am. Chem. Soc. 142 (2020) 13041–13050; (g) L. Fan, J. Hao, J. Yu, et al., J. Am. Chem. Soc. 142 (2020) 6698–6707; (h) L. Kong, X. Han, S. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 7188–7192; (i) Y. Luo, H. Liu, J. Zhang, M. Liu, L. Dong, Org. Lett. 22 (2020) 7604–7608; (j) H. Li, X. Cui, Chin. J. Org. Chem. 2 (2020) 543–544; (k) Y. Dong, R. Liu, W. Wang, Green Synth. Catal. 2 (2020) 83–85. |
| [10] |
(a) J. Wu, X. Cui, L. Chen, G. Jiang, Y. Wu, J. Am. Chem. Soc. 131 (2009) 13888–13889; (b) X.X. Guo, D.W. Gu, Z. Wu, W. Zhang, Chem. Rev. 115 (2015) 1622–1651; (c) F. Wang, S. Yu, X. Li, Chem. Soc. Rev. 45 (2016) 6462–6477; (d) X. Wang, A. Lerchen, T. Gensch, et al., Angew. Chem. Int. Ed. 56 (2017) 1381–1384; (e) Y. Zhao, S. Li, X. Zheng, et al., Angew. Chem. Int. Ed. 56 (2017) 4286–4289; (f) Y. Li, F. Xie, Y. Liu, X. Yang, X. Li, Org. Lett. 20 (2018) 437–440. |
| [11] |
(a) G. Brasche, S.L. Buchwald, Angew. Chem. Int. Ed 47 (2008) 1932–1934; (b) B. Ma, Y. Wang, J. Peng, Q. Zhu, J. Org. Chem. 76 (2011) 6362–6366; (c) J. Li, L. Neuville, Org. Lett. 15 (2013) 1752–1755; (d) J. Li, M. John, L. Ackermann, Chem. Eur. J. 20 (2014) 5403–5408; (e) Y. Zhu, C. Li, J. Zhang, et al., Org. Lett. 17 (2015) 3872–3975; (f) J. Li, M. Tang, L. Zang, et al., Org. Lett. 18 (2016) 2742–2745; (g) X. Zhou, Z. Qi, S. Yu, et al., Adv. Syn. Catal. 359 (2017) 1620–1625; (h) J. Zhou, J. Li, Y. Li, et al., Org. Lett. 20 (2018) 7645–7649; (i) H.B. Xu, Y.Y. Zhu, L. Dong, J. Org. Chem. 84 (2019) 16286–16292; (j) H. Xing, J. Chen, Y. Shi, et al., Org. Chem. Front. 7 (2020) 672–677; (k) F. Xu, Y.Y. Song, W.J. Zhu, et al., Chem. Commun. 56 (2020) 11227–11230. |
| [12] |
(a) Y. Li, Z. Qi, H. Wang, X. Yang, X. Li, Angew. Chem. Int. Ed. 55 (2016) 11877–11881; (b) Z. Qi, S. Yu, X. Li, Org. Lett. 18 (2016) 700–703; (c) R. Lai, X. Wu, S. Lv, et al., Chem. Commun. 55 (2019) 4039–4042. |
| [13] |
(a) L. Xu, L. Wang, Y. Feng, et al., Org. Lett. 19 (2017) 4343–4346; (b) Y. Li, C. Jia, H. Li, et al., Org. Lett. 20 (2018) 4930–4933; (c) S. Huang, H. Li, X. Sun, et al., Org. Lett. 21 (2019) 5570–5574; (d) Y. Hu, T. Wang, Y. Liu, et al., Org. Lett. 22 (2019) 501–504. |
| [14] |
(a) X. Wei, M. Zhao, Z. Du, X. Li, Org. Lett. 13 (2011) 4636–4639; (b) G. Zheng, M. Tian, Y. Xu, X. Chen, X. Li, Org. Chem. Front. 5 (2018) 998–1002; (c) Q. Yang, C. Wu, J. Zhou, et al., Org. Chem. Front. 6 (2019) 393–398; (d) F. Xu, Y.Y. Song, W.J. Zhu, et al., Chem. Commun. 56 (2020) 11227–11230. |
| [15] |
(a) C. You, C. Pi, Y. Wu, X. Cui, Adv. Syn. Catal. 360 (2018) 4068–4072; (b) S. Du, C. Pi, T. Wan, Y. Wu, X. Cui, Adv. Syn. Catal. 361 (2019) 1766–1770; (c) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374–1378; (d) T. Yuan, C. Pi, C. You, et al., Chem. Commun 55 (2019) 163–166; (e) J. Ren, C. Pi, Y. Wu, X. Cui, Org. Lett. 21 (2019) 4067–4071; (f) Y. Wu, C. Pi, X. Cui, Y. Wu, Org. Lett. 22 (2020) 361–364; (g) J. Ren, X. Yan, X. Cui, et al., Green Chem. 22 (2020) 265–269. |
| [16] |
Q. Mei, L. Wang, Y. Guo, et al., J. Mater. Chem. 22 (2012) 6878-6884. |
| [17] |
(a) R. Rohini, K. Shanker, P.M. Reddy, Y.P. Ho, V. Ravinder, Eur. J. Med. Chem. 44 (2009) 3330–3339; (b) J.C. Li, R.X. Wang, Y. Sun, et al., Bioorgan. Chem. 92 (2019) 103266. |