 2021, Vol. 32
2021, Vol. 32 


Selenenylated molecules have widespread applications in the fields of pharmaceuticals and materials science because of their interesting biological activities [1] and suitable fluorescence properties [2]. Furthermore, organoselenium compounds have been widely used as convenient intermediates or reagents and catalysts in organic synthesis [3]. Thus, significant efforts have been made to develop various strategies for the incorporation of selenium functional groups into organic molecules [4].
On the other hand, the difunctionalization of readily accessible alkenes has become a powerful tool for the construction of highly functionalized skeletons in organic synthesis [5]. In this context, selenocyclizations of unsaturated amines are a useful concept for the preparation of selenenylated and nitrogenated heterocycles, which could provide an excellent opportunity for medicinal chemists in drug discovery. Additionally, the simultaneous incorporation of synthetically versatile seleno and nitro moieties has enabled further elaboration of these products as intermediates in syntheses of structurally diverse molecules. Accordingly, over the years, substantial efforts have been devoted to advance this topic, and numerous elegant procedures have been developed as a result. Early reports on these reactions usually involved electrophilic selenium species-induced cyclizations with organoselenium reagents, e.g., phenylselenenyl halides have been reported with olefinic amines [6], (sulfon)amides [7], oximes [8], thiazolines [9], carbamates [10], imines [11], imidates [12], hydrazines [13] and hydrazine [14] (Scheme 1a). Typically, this selenoamination of olefins occurs via intramolecular nucleophilic attack onto seleniranium ion 3 to form nitrogen-containing heterocycles with the aid of additives, such as Lewis acids [8a], Brønsted acids [7a, 13], or silica gel [7e, 10b–10e]. However, the presence of nucleophilic halides often causes some undesirable processes, such as the incorporation of halogens, which decrease the selectivity [15]. Additionally, the addition adducts sometimes can further react with phenylselenenyl halides to afford the deselenenylated products [16]. Furthermore, these procedures are usually incompatible with a large number of important functional groups due to the formation of hydrogen halide as a side-product during the course of the reaction.

|
Download:
|
| Scheme 1. Intramolecular selenoamination. | |
Diselenide is preferable as an alternative selenylation agent in this transformation because it is bench-stable, readily available and easy to handle during the synthesis process. For example, Tiecco et al. reported the reaction of unsaturated amines with phenylselenenyl sulfate, generated from the oxidation of diphenyl diselenide with ammonium persulfate in the presence of trifluoromethanesulfonic acid, to afford phenylseleno-substituted N-heterocyclics (Scheme 1b) [17]. Again, these cyclization reactions were carried out in acidic conditions, and substrates bearing acid-sensitive functional group, such as silyl ethers or ketones, were not tolerated under these reaction conditions.
Despite these advances, general methods for the preparation of various arylselenopyrrolidines and alkylselenopyrrolidines in particular are still limited. Our goal was to develop an efficient and operationally simple selenoamination, in which readily available, inexpensive, and easy-to-handle reagents were employed. In continuation of our interest in hypervalent iodine chemistry [18], we conjectured that diselenides would be oxidized in the presence of a commercially available hypervalent iodine(III) reagent to form electrophilic selenium species (RSe+), which would participate in the electrophilic selenocyclization of olefins (Scheme 1c).
We started our investigation with the optimization of the reaction conditions using N-(2, 2-diphenylpent-4-en-1-yl)-4-methylbenzenesulfonamide (1a) as a model substrate due to its ease of preparation and UV-absorption property, which facilitates the monitoring of the reaction progress. The preliminary results are summarized in Table 1.
|
|
Table 1 Optimization of the reaction conditions. a |

{kind=link}
Based on our previous results [18a], we first employed iodine pentoxide (I2O5) as reaction promoter and diphenyl diselenide (Ph2Se2) (2a) as selenium source. However, only a trace amount of selenoamination product 3a was obtained (Table 1, entry 1). Other promoters, such as oxone, K2S2O8, O2, PhI(OAc)2 and PhIO were tested to further improve the yield, and PhIO produced the best result (entries 2–6). The effect of the solvents on the course of the reaction was also noteworthy (entries 7–10), and CH 2Cl2 was the most suitable solvent for the reaction. Decreasing the amount of Ph2Se2 to 0.5 equiv. led to a decrease in the conversion (entry 11). Control experiments indicated that PhIO played an important role in this transformation, as no reaction occurred in the absence of PhIO (entry 12).
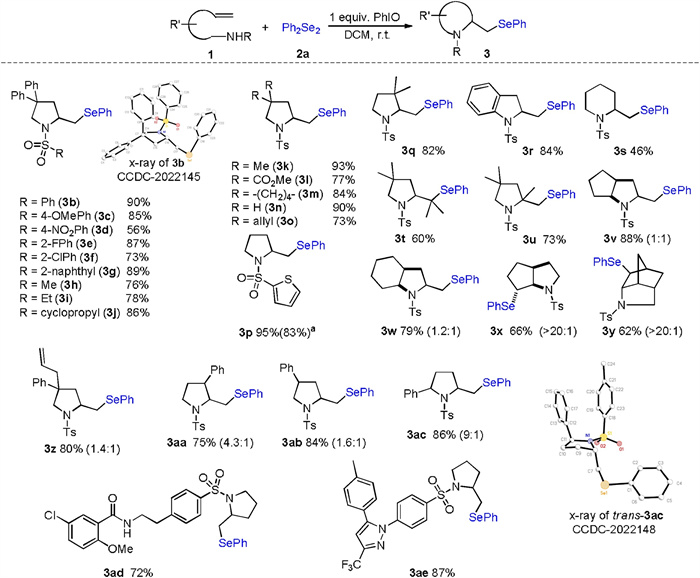
With these optimized reaction conditions, we turned our attention to investigating the substrate scope of the title transformation with a diverse range of N-alkenyl sulfonamides using 2a as the selenium source. As indicated in Scheme 2, both N-aryl and N-alkylsulfonamides produced the corresponding 2-phenylselenomethylpyrrolidines with good to excellent isolated yields (3b-3j). The structural skeleton of 2-phenylselenomethylpyrrolidine was confirmed by an X-ray diffraction experiment performed with compound 3b (CCDC: 2022145). As shown in the ORTEP drawing, 3b bore an envelope chair conformation with two phenyl groups at both axial and equatorial positions, the phenylseleno moiety and Ts group also stayed at equatorial positions. Notably, p-nitrobenzenesulfonamide resulted in the corresponding pyrrolidine derivative 3d with moderate yield. This is most likely because of the low nucleophilicity of sulfonamide caused by the strong electron-withdrawing nitro group at the para position of the benzene ring. Reactions of substrates bearing different substituents on the main chain proceeded well to produce their corresponding products 3k–3q with good yields. It is noteworthy that the Thorpe–Ingold effect was not required in the current cyclization, and selenoaminated products3n and 3p were isolated with 90% and 95% yields, respectively.

|
Download:
|
| Scheme 2. Scope of the intramolecular selenoamination. Reaction conditions: 1 (0.2 mmol, 1.00 equiv.), 2a (0.2 mmol, 1.00 equiv.), PhIO (0.2 mmol, 1.00 equiv.), DCM (2 mL), r.t., the numbers in the parentheses are the diastereomeric ratios determined by 1H NMR. a 5 mmol scale. | |
{kind=link}
Importantly, the protocol of the present reaction was very simple: The alkene, PhIO and diselenide were stirred in simple glassware under ambient conditions. Thus, this reaction could be applied to a large-scale synthesis without any difficulties. For instance, when the reaction of 1p (1.16 g, 5 mmol) was performed with Ph2Se2 (1.56 g, 5 mmol) and PhIO (1.10 g, 5 mmol) under the optimized reaction conditions, the desired product 3p was obtained with 83% yield (1.60 g) after recrystallization.
N-Tosyl o-allyl aniline 1r worked well in this transformation, and the corresponding indoline 3r was obtained with good yield. Reaction of 1-sulfonamido-5-hexene substrate gave lower isolated yield of seleno piperidine (3s), although a lower yield was obtained, possibly due to unfavorable entropy of the current cyclization [19]. Substituents on the C=C double bonds showed a low impact on the reaction. Di- and trisubstituted terminal alkenes, such as 1t and 1u, were efficiently converted into their corresponding pyrrolidines with satisfactory yields. More complex bicyclic and bridged ring systems were also accessible using this protocol (3v-3y). The relative configuration of the phenylseleno unit and amino group in 3x and 3y was assigned to trans, as expected for a selenoamination via an intermediate seleniranium ion. The desymmetrization of diene 1z delivered seleno-pyrrolidine 3z with 80% yield as a mixture of diastereomers. Substrates bearing one substitute on the main chain also underwent high cyclization to produce the corresponding pyrrolidines 3aa-3ac with a good isolated yield but low diastereoselectivity. The resulting disubstituted pyrrolidines can serve as useful precursors for the synthesis of biological hexahydro-1H-pyrrolizines and octahydroindolizines [20]. The relative stereochemistry of trans-3ac was established by an X-ray diffraction experiment (CCDC: 2022148). Finally, we further applied this methodology to architecturally complex sulfonamide substrates. In this regard, a glibenclamide precursor-derived sulfonamide smoothly engaged in this selenoamination and afforded acceptable yields of 3ad. The sulfonamide 1ae derived from celecoxib, which is a nonsteroidal anti-inflammatory drug, worked well under the standard conditions, delivering the corresponding products 3ae with good yields.
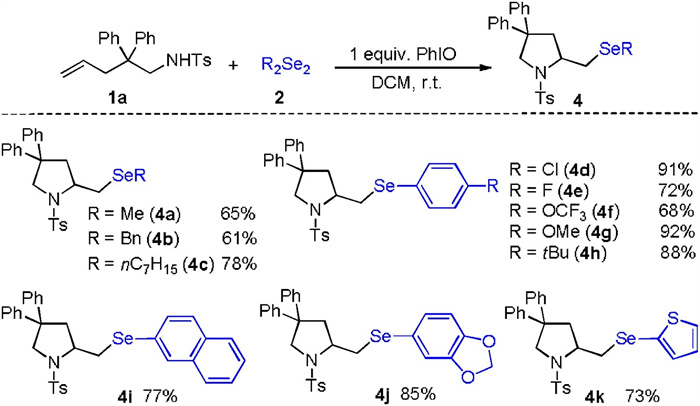
Next, we examined the generality of this transformation with respect to different diselenides, and the results are summarized in Scheme 3. As depicted in Scheme 3, the dialkyl diselenides were suitable reaction partners under the reaction conditions, and provided the corresponding products with good yields (4a-4c). Thus, this newly developed PhIO-promoted selenoamination provides, for the first time, a feasible way to access alkylselenopyrrolidines. Different diaryl diselenides with an electron-withdrawing group or electron-donating group at the para position all showed good reactivity in the procedure (4d-4h). Additionally, naphthyl diselenide was successfully reacted with 1a to produce the respective 2-selenomethylpyrrolidine 4i with 77% yield. Finally, heteroaryl diselenides, such as dithienyl diselane were subjected to the procedure, and the selenation product (4k) was isolated with 73% yields. Unfortunately, other dichalcogenides, such as diphenyl disulfide (Ph2S2) and diphenyl ditelluride (Ph2Te2), failed to produce the desired products in this transformation, presumably because Ph2S2 and Ph2Te2 cannot be oxidized to the corresponding electrophilic species under the present conditions.

|
Download:
|
| Scheme 3. Scope of diselenides. Reaction conditions: 1a (0.2 mmol, 1.00 equiv.), 2 (0.2 mmol, 1.00 equiv.), PhIO (0.2 mmol, 1.00 equiv.), DCM (2 mL), r.t. | |
{kind=link}
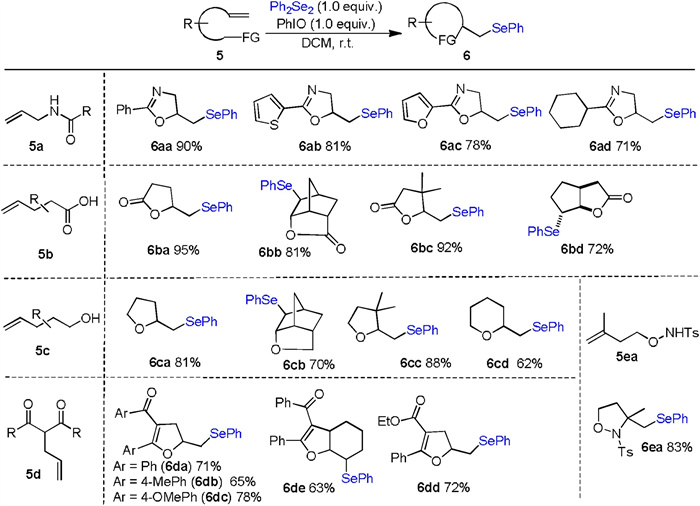
In consideration of the remarkably broad substrate scope accessible from this transformation, the current procedure was applied to the preparation of other seleno-containing heterocycles. As shown in Scheme 4, a series of seleno oxazolines were produced successfully under optimal conditions (6aa-6ad). The generality of the title procedure has also been demonstrated by a series of unsaturated carboxylic acids furnishing the corresponding phenylselenolactones in good yield (6ba-6bd). Additionally, using the alkenoic alcohols as substrates, we observed the construction of five- and six-membered cyclic ethers in high yields (6ca-6cd). Subsequently, we continued to investigate the generality of this transformation with respect to different olefinic dicarbonyl compounds. As we expected, different seleno dihydrofurans (6da-6de) could be obtained in acceptable isolated yields under standard conditions. Finally, preparation of isoxazolidine 6ea was also possible using N-alkenoxyl sulfonamide derivative 5ea as the starting material.

|
Download:
|
| Scheme 4. Selenocyclization of different olefins. Reaction conditions: 5 (0.2 mmol, 1.00 equiv.), Ph2Se2 (0.2 mmol, 1.00 equiv.), PhIO (0.2 mmol, 1.00 equiv.), DCM (2 mL), r.t. | |
{kind=link}
Mechanistic studies were then performed to gain knowledge regarding the reaction pathway. Initially, no dramatic decline in the yield was observed after the addition of 2.0 equiv. of 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO) to the reaction mixture (Scheme 5a), which indicated that a radical process likely did not occur. PhIO was insoluble in CH2Cl2 because it displays a zigzag polymeric chain structure according to X-ray powder diffraction and EXAFS studies [21]. It, however, could be mostly dissolved when Ph2Se2 was added to the CH2Cl2 solution (Fig. S1 in Supporting information). This result indicated that PhIO can react with Ph2Se2 to generate new reactive species in the current reaction system. This phenomenon was in accordance with NMR observations, which showed that the signal pattern for the aromatic protons of Ph2Se2 changed significantly after the addition of PhIO, indicating the mutual interaction between Ph2Se2 and PhIO and, thus, resulting in changes in the electronic environment surrounding the phenyl groups in Ph2Se2 (Fig. S2 in Supporting information).

|
Download:
|
| Scheme 5. Experiments for mechanistic studies. | |
{kind=link}
When the reaction was performed in acetone under otherwise identical conditions, α-phenylselenenylated ketone 7 was obtained unexpectedly instead of the selenoamination product (Scheme 5b). Compound 7 was probably a result of an electrophilic attack of the phenylselenenyl cation (PhSe+) to the enol form of acetone [22]. Furthermore, 2 equiv. of indole was added to the reaction mixture under optimal conditions to capture any possible PhSe+ formed during the reaction. Indole was employed as a competitive substrate based on the fact that it could be easily selenenylated by PhSe+ [23]. As expected, indolyl selenide8 was detected with 15% isolated yield (Scheme 5c).
According to the aforementioned information and previous reports [24], a possible reaction pathway for this transformation is outlined in Scheme 6. First, diselenide is oxidized by PhIO to generate RSe+, which serves as a reactive electrophilic intermediate to induce the title selenocyclization.

|
Download:
|
| Scheme 6. Tentative reaction pathway. | |
{kind=link}
In summary, we developed an iodosobenzene-mediated intramolecular selenocyclization of olefins, which produced selenenylated heterocycles with good yields and excellent functional group compatibility. Preliminary mechanistic studies indicate that the title reaction takes place via an active electrophilic selenium intermediate generated in situ from PhIO and diselenide. We believe that the developed methodology can be applied to potential operational procedures for the synthesis of nitrogen- and selenium-containing molecules due to its good isolated yields, high functional group tolerance, mild conditions and operational simplicity. Efforts are in progress to elucidate the mechanistic details of this reaction by means of DFT-calculations and to disclose its scope and limitations.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis study was supported by the Natural Science Foundation of Jiangsu Province (No. BK20170439), Science and Technology Plan Projects of Nantong (Nos. JC2019102 and JC2020072).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.050.
| [1] |
(a) Z. Chen, H. Lai, L. Hou, T. Chen, Chem. Commun. 56 (2020) 179–196; (b) C.W. Nogueira, G. Zeni, J.B.T. Rocha, Chem. Rev. 104 (2004) 6255–6285; (c) G. Mugesh, W.W. du Mont, H. Sies, Chem. Rev. 101 (2001) 2125–2179. |
| [2] |
(a) S. Panda, A. Panda, S.S. Zade, Coord. Chem. Rev. 300 (2015) 86–100; (b) S.T. Manjare, Y. Kim, D.G. Churchill, Acc. Chem. Res. 47 (2014) 2985–2998. |
| [3] |
(a) A. Breder, C. Depken, Angew. Chem. Int. Ed. 58 (2019) 17130–17147; (b) J. Luo, X. Liu, X. Zhao, Synlett 28 (2017) 397–401; (c) V. Rathore, C. Jose, S. Kumar, New J. Chem. 43 (2019) 8852–8864; (d) P.N. Makhal, A. Nandi, V.R. Kaki, ChemistrySelect 6 (2021) 663–679. |
| [4] |
(a) A.D. Sonawane, M. Koketsu, Curr. Org. Chem. 23 (2019) 3206–3225; (b) A.J. Mukherjee, S.S. Zade, H.B. Singh, R.B. Sunoj, Chem. Rev. 110 (2010) 4357–4416; (c) K. Sun, X. Wang, C. Li, H. Wang, L. Li, Org. Chem. Front. 7 (2020) 3100–3119. |
| [5] |
(a) X.W. Lan, N.X. Wang, Y. Xing, Eur. J. Org. Chem. 2017 (2017) 5821–5851; (b) T. Courant, G. Masson, J. Org. Chem. 81 (2016) 6945–6952; (c) Z.L. Li, G.C. Fang, Q.S. Gu, X.Y. Liu, Chem. Soc. Rev. 49 (2020) 32–48; (d) J.S. Zhang, L. Liu, T. Chen, L.B. Han, Chem. Asian J. 13 (2018) 2277–2291. |
| [6] |
(a) X. Franck, S. Leleu, F. Outurquin, Tetrahedron Lett. 51 (2010) 4437–4440; (b) B. Berthe, F. Outurquin, C. Paulmier, Tetrahedron Lett. 38 (1997) 1393–1396. |
| [7] |
(a) T. Izumi, M. Sugano, T. Konno, J. Heterocycl. Chem. 29 (1992) 899–904; (b) A.D. Jones, D.W. Knight, A.L. Redfern, J. Gilmore, Tetrahedron Lett. 40 (1999) 3267–3270; (c) A.D. Jones, A.L. Redfern, D.W. Knight, I.R. Morgan, A.C. Williams, Tetrahedron 62 (2006) 9247–9257; (d) A. Toshimitsu, K. Terao, S. Uemura, Tetrahedron Lett. 25 (1984) 5917–5920; (e) A. Toshimitsu, K. Terao, S. Uemura, J. Org. Chem. 51 (1986) 1724–1729. |
| [8] |
(a) R. Grigg, M. Hadjisoteriou, P. Kennewell, J. Markandu, J. Chem. Soc. Chem. Commun. (1992) 1537–1538; (b) M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli, J. Chem. Soc. Chem. Commun. (1995) 235–236. |
| [9] |
K. Terao, M. Kunishima, S. Tani, Synlett 6 (1999) 733-734. DOI:10.1055/s-1999-2728 |
| [10] |
(a) R.R. Webb II, S. Danishefsky, Tetrahedron Lett. 24 (1983) 1357–1360; (b) M.A. Cooper, A.D. Ward, Tetrahedron Lett. 33 (1992) 5999–6002; (c) A.M. Morella, A.D. Ward, Aust. J. Chem. 48 (1995) 445–467; (d) D.L.J. Clive, C.K. Wong, W.A. Kiel, S.M. Menchen, J. Chem. Soc. Chem. Commun. (1978) 379–380; (e) D.L.J. Clive, V. Farina, A. Singh, et al., J. Org. Chem. 45 (1980) 2120–2126. |
| [11] |
(a) N. De Kimpe, M. Boelens, J. Chem. Soc. Chem. Commun. (1993) 916–918; (b) D. Schley, J. Liebscher, Eur. J. Org. Chem. 2007 (2007) 2945–2957. |
| [12] |
(a) K. Terao, A. Toshimitsu, S. Uemura, J. Chem. Soc. Perkin Trans. 1 (1986) 1837–18431709c(b) A. Toshimitsu, K. Terao, S. Uemura, J. Chem. Soc. Chem. Commun. (1986) 530–531.
|
| [13] |
M. Tiecco, L. Testaferri, F. Marini, et al., Tetrahedron 53 (1997) 10591-10602. DOI:10.1016/S0040-4020(97)00670-4 |
| [14] |
M. Tiecco, L. Testaferri, F. Marini, et al., Tetrahedron 53 (1997) 7311-7318. DOI:10.1016/S0040-4020(97)00414-6 |
| [15] |
(a) X. Pannecoucke, F. Outurquin, C. Paulmier, Eur. J. Org. Chem. 2002 (2002) 995–1006; (b) F. Outurquin, X. Pannecoucke, B. Berthe, C. Paulmier, Eur. J. Org. Chem. 2002 (2002) 1007–1014. |
| [16] |
(a) M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli, D. Bartoli, Tetrahedron 44 (1988) 2261–2272: (b) M. Tiecco, L. Testaferri, M. Tingoli, D. Chianelli, D. Bartoli, Tetrahedron 44 (1988) 2273–2282. |
| [17] |
(a) M. Tiecco, L. Testaferri, F. Marini, et al., Tetrahedron 53 (1997) 4441–4446; (b) M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli, F. Marini, J. Chem. Soc. Perkin Trans. 1 (1993) 1989–1993; (c) M. Tiecco, L. Testaferri, F. Marini, Tetrahedron 52 (1996) 11841–11848; (d) M. Tiecco, L. Testaferri, M. Tingoli, C. Santi, Tetrahedron Lett. 36 (1995) 163–166. |
| [18] |
(a) W. Yi, P.F. Wang, M. Lu, et al., ACS Sustain. Chem. Eng. 7 (2019) 16777–16785; (b) W. Yi, Q.Y. Liu, X.X. Fang, S.C. Lou, G.Q. Liu, Org. Biomol. Chem. 16 (2018) 7012–7018; (c) J. Liu, Q.Y. Liu, X.X. Fang, G.Q. Liu, Y. Ling, Org. Biomol. Chem. 16 (2018) 7454–7460; (d) G.Q. Liu, Y.M. Li, J. Org. Chem. 79 (2014) 10094–10109. |
| [19] |
(a) G. Illuminati, L. Mandolini, Acc. Chem. Res. 14 (1981) 95–102; (b) A.J. Kirby, Effective Molarities for Intramolecular Reactions, Vol. 17, Academic Press, 1980, pp. 183–278; (c) C. Galli, L. Mandolini, Eur. J. Org. Chem. 2000 (2000) 3117–3125. |
| [20] |
M. Tiecco, L. Testaferri, L. Bagnoli, C. Scarponi, Tetrahedron: Asymmetry 19 (2008) 2411-2416. DOI:10.1016/j.tetasy.2008.10.006 |
| [21] |
C.J. Carmalt, J.G. Crossley, J.G. Knight, et al., J. Chem. Soc. Chem. Commun. (1994) 2367-2368. |
| [22] |
N. Miyoshi, T. Yamamoto, N. Kambe, S. Murai, N. Sonoda, Tetrahedron Lett. 23 (1982) 4813-4816. DOI:10.1016/S0040-4039(00)85720-2 |
| [23] |
(a) Q.B. Zhang, Y.L. Ban, P.F. Yuan, et al., Green Chem. 19 (2017) 5559–5563; (b) V. Rathore, S. Kumar, Green Chem. 21 (2019) 2670–2676. |
| [24] |
(a) M. Shi, B.Y. Wang, J. Li, Eur. J. Org. Chem. 2005 (2005) 759–765; (b) M. Tiecco, M. Tingoli, L. Testaferri, Pure Appl. Chem. 65 (1993) 715–722; (c) L. Yu, B. Chen, X. Huang, Tetrahedron Lett. 48 (2007) 925–927. |