 2021, Vol. 32
2021, Vol. 32 


Nitrogen-containing heterocycles are ubiquitous structural units that appear in numerous naturally occurring products, pharmaceuticals, and functional materials, among others. Representatively, indoles and carbazoles are recognized as privileged architectures in biologically active compounds [1] and organic optoelectronic materials [2], respectively. Consequently, intensive attention has been continuously drawing upon their synthetic methodology [3].
While some traditional name reactions provide viable access to these nitrogen heterocycles [4], in the last few decades, diverse strategies such as radical couplings [5], nitrene insertion [6], and transition-metal-catalyzed C–H amination [7] have been widely exploited to bring about bond formation/N-heterocyclization. Specifically, in qualitative complementary to the Sundberg method [8] for carbazole/indole formation, Cadogan reaction [9] represents an essential alternative for azacyclizations from stable and easily accessible feedstock reagents and has widespread applications in the preparation of carbazole-based functional materials (Scheme 1a). The common Cadogan transformations utilize stoichiometric amounts of phosphines at very high temperatures (150−220 ℃) [10]. Milder catalytic protocols (80−120 ℃) based on transition-metal catalysis have also been developed [11]. Intriguingly, Radosevich et al. recently designed a novel small-ring phosphacycloalkane that, combined with hydrosilane as the terminal electron acceptor, served as an efficient catalyst to enable scalable Cadogan-type azacyclizations with both C-N and N-N bond formation [12]. Despite utilities, these methods still suffer from certain restrictions such as the indispensable transition metal catalysis, air-sensitive reagent, or thermal conditions at high temperature; thereby the development of mild and user-friendly strategies for the formation of carbazoles and related heterocycles remains highly desirable.

|
Download:
|
| Scheme 1. Carbazole formation via intramolecular reductive amination of o-nitrobiphenyl. | |
Photoredox catalysis provides an attractive alternative approach for the valuable N-heterocycle synthesis [13], with yet few reports on carbazole construction using sustainable visible light [14]. With primary insight into photolysis heterocyclization of o-azido compounds [15] and recent photoredox reductive couplings [16], we speculate that photo-irradiation could activate the proposed intermediate of Cadogan reaction in the rate-determining deoxygenation process [9c], and thereby break the thermodynamic barriers inherent to the overall cyclization under mild conditions (Scheme 1b). Moreover, the electronically opposite properties of nitro biaryls and phosphines may lead to the transient generation of photosensitive electron donor-acceptor (EDA) complexes [17], thus probably averting the need for external photosensitizer. With our recent effort on this subject, herein, we describe the mild visible-light-mediated Cadogan reaction, which unexpectedly could be dramatically enhanced by an organic photocatalyst. Hence, the photoactivation process of the vital deoxygenation process in this protocol has been discovered.
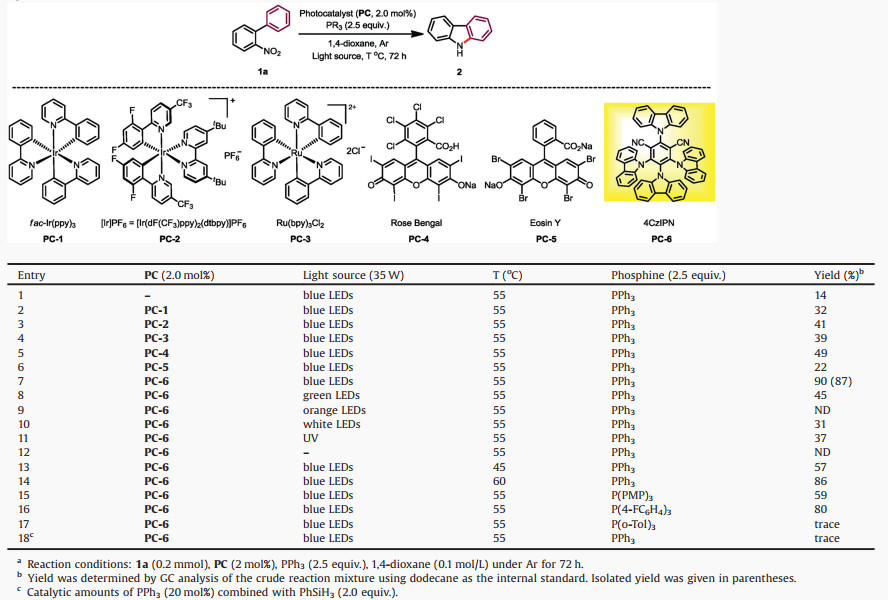
To commence our studies, we set up the Cadogan reaction by choosing o-nitrobiphenyl (1a) as the model substrate to optimize reaction conditions (Tables S1-S8 in Supporting information for details) and key parameters of screening the photoredox Cadogan reaction are shown in Table 1. The photochemical reaction of o-nitrobiphenyl 1a was initially treated with stoichiometric triphenylphosphine (PPh3) in the absence of any photosensitizer or other additive. Visible light stimulation upon this mixture with blue LEDs led to the formation of carbazole 2 in 14% yield within three days, with the majority of starting material and PPh3 recovered (Table 1, entry 1). In order to improve the efficiency of this cyclization transformation, a pad of photocatalysts were screened. Among them, Ir or Ru complexes (PC-1 to PC-3) enhanced the cyclization to afford product in modest yields (Table 1, entries 2–4). Then, organophotocatalysts were used. While Rose Bengal featured moderate catalytic activity (49% yield, Table 1, entry 5), Eosin type photosensitizer gave a slightly increased yield than the catalyst-free conditions (22% yield, Table 1, entry 6). To our delight, the organic D-A type photosensitizer 4CzIPN was superior to others, furnishing the carbazole product in excellent yield with full conversion of 1a (90% yield, Table 1, entry 7). After screening different light sources (Table 1, entries 8–12), no desired product was detected when using orange LEDs or under the dark conditions. Further, green LEDs and white LEDs could enable this reaction, albeit in low yields. Notably, the reaction temperature dramatically affected the efficiency of reaction systems, where mild thermal condition was required (50−60 ℃) (Table 1, entries 13–14). Regarding phosphine reductants (Table 1, entries 15–17), among others P(4-FC6H4)3 gave a good result with 4CzIPN. Tris(4-methoxyphenyl)phosphine [P(PMP)3] and tricyclohexylphosphine [P(Cy)3] featured moderate reactivities (Table S3 in Supporting information for details). We also used PPh3 in a catalytic amount combined with stoichiometric PhSiH3 (Table 1, entry 18), which generated 2-phenylaniline as the major product.
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
With the catalytic capacity of 4CzIPN for the mild photochemcial Cadogan cyclization, we next probed the substrate scope of the current system. Generally, the 4CzIPN-based photochemical system featured high reactivities to afford the carbazole products in good to excellent yields (Scheme 2). A broad range of functionalities attached at the o-nitrobiphenyl motif such as methoxy (5), trifluoromethyl (6), halogen (7, 17), nitrile (8), acetyl (9, 15), ester (10), amide (11), formyl (12), free hydroxymethyl (13), and even alkenyl (14) were all smoothly accommodated (40%–89% yields). Among them, the substrates with electron-deficient groups generally found higher reactivity than those bearing electron-donating substituents, which in turn required prolonged reaction times. The model reaction on a gram-scale also proved highly effective and full conversion was observed within 4 d by the using two 35 W blue LEDs (7.5 mmol scale, 76% yield). Notably, the reaction could complete within 24 h when increasing catalyst loading to 10 mol%.

|
Download:
|
| Scheme 2. Substrate scope for carbazole formation. a 10 mol% of photocatalyst was used within 24 h. b Major isomer (rr=3:1 determined by GC). c On a 0.1 mmol scale. | |
{kind=link}
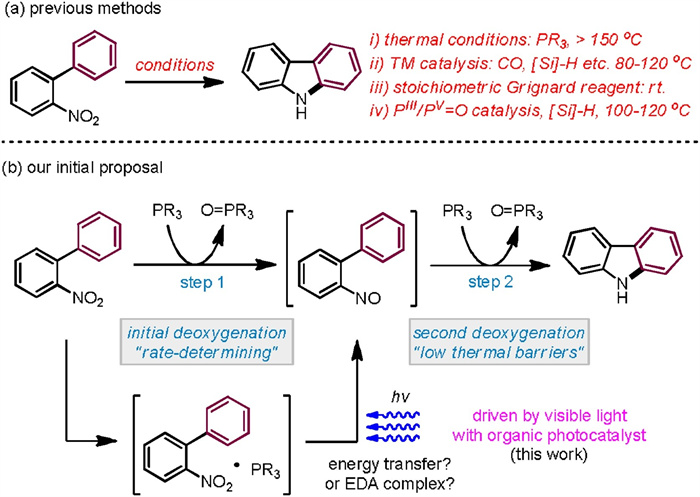
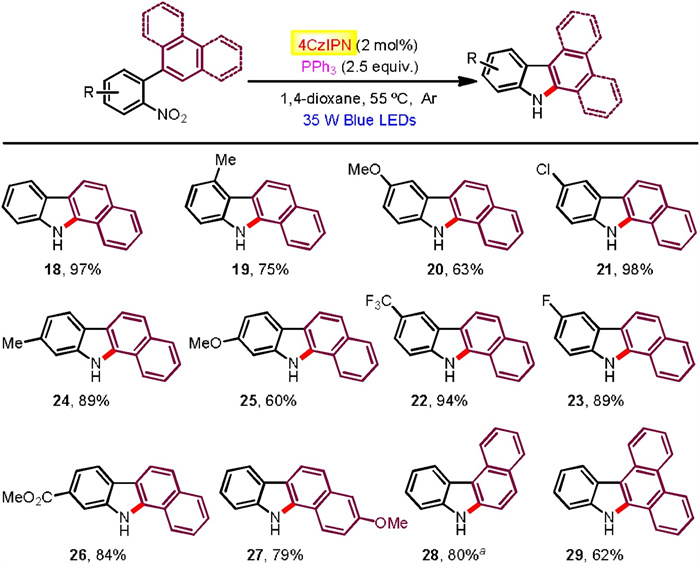
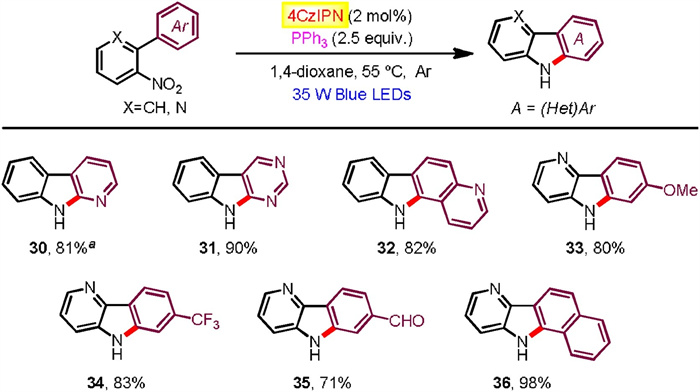
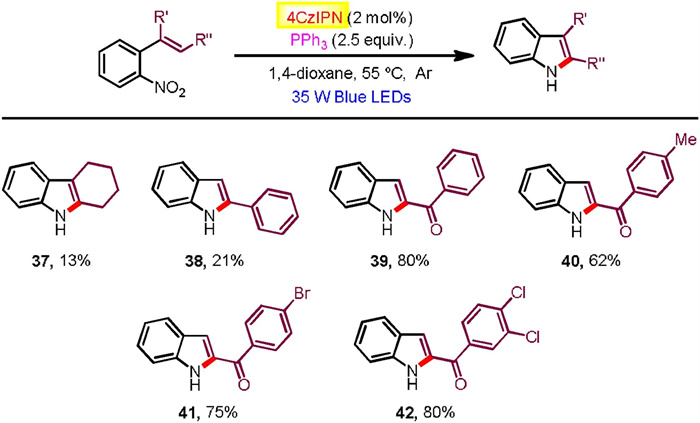
Moreover, 2-(2-nitrophenyl)naphthalene substrates found generally high reactivities and exclusive regioseletivities (Scheme 3). Hence, a number of benzo[a]carbazoles have been accessed in good to excellent yields (60%–98% yields), tolerating a pad of useful functional groups attached at the nitrophenyl moiety (18-27). Additionally, 1-(2-nitrophenyl)naphthalene and 9-(2-nitrophenyl)phenanthrene were successfully participated to afford the corresponding benzo[c]carbazole (28) and dibenzo[a, c]carbazole (29), respectively. We also exploited aza-biheteroarene substrates such as those with pyridinyl (30, 33-36), pyrimidinyl (31), and quinolinyl (32) moieties (Scheme 4). All of them exhibited good efficiency to furnish azacarbazole and azabenzocarbazole products (71%–98% yields). In terms of o-nitrostyrene reactants (37-42), they worked with effectiveness highly dependent on the electron effect of substituents (Scheme 5). Among them, electron withdrawing acyl groups (enones) delivered 2-acylindole products with satisfactory conversions (39-42, 62%–80% yields). Majority of reactants were recovered in the cases of substrates containing cyclohexanene and stilbene moieties. We also tried to improve the reactivity of these unactivated alkenes by modification of reaction conditions including solvent, photocatalyst loading and reaction time, but the yields of indoles37 and 38 did not increase significantly.

|
Download:
|
| Scheme 3. Substrate scope for benzocarbazole formation. a On a 0.1 mmol scale. | |
{kind=link}

|
Download:
|
| Scheme 4. Substrate scope for azacarbazole formation. a Major isomer (rr=10:1 determined by GC). | |
{kind=link}

|
Download:
|
| Scheme 5. Substrate scope for indole formation. | |
{kind=link}
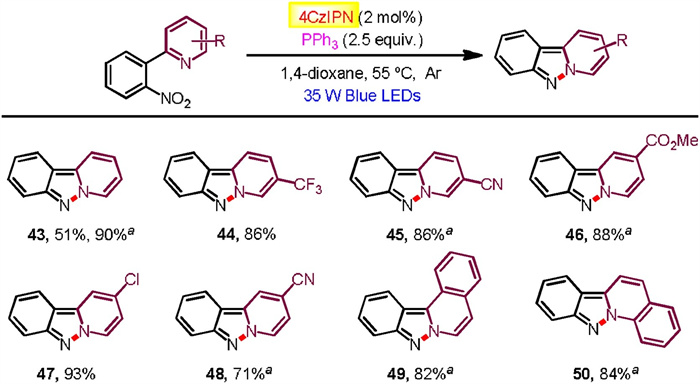
The robust nature of our photocatalytic systems for Cadogan cyclization was further mirrored by the effective N-N bond formation (Scheme 6). Thereby, some functionalized 2-(2-nitrophenyl)pyridines transformed to the corresponding pyrido[1, 2-b]indazoles in high yields (43-48, 71%–90% yields), as well as 1-(2-nitrophenyl)isoquinoline (49, 82% yield) and 2-(2-nitrophenyl)quinoline (50, 84% yield). It is noted worth that there appears a substantial reactivity enhancement with smaller reaction scales in some cases that the starting materials did not convert completely on a larger scale even altering the reaction conditions with higher concentration.

|
Download:
|
| Scheme 6. Substrate scope for pyrido[1, 2-b]indazole formation. a On a 0.1 mmol scale. | |
{kind=link}
With the established reactivities of our mild visible-light-driven Cadogan cyclization, we were attracted to depict its photoactivation model. To this end, some mechanistic experiments were carried out. First, the fluorescence quenching experiments disclosed that while both 2-nitrobiphenyl (1a) and PPh3 displayed Stern-Volmer quenching effect with KSV to be 354 and 105, respectively [18], the combination of them obviously enhanced such effect upon the excited photocatalyst (KSV=670), suggesting formation of a complex of 1a and PPh3 and superior energy transfer from the excited 4CzIPN to this complex (Scheme 7a) [19]. Then, the independent and competition kinetic isotope effect (KIE) of carbazole formation was found to bekH/kD = 1.0 and 1.1, respectively, indicating a kinetically irrelevant C–H cleavage in the photochemical Cadogan cyclization (Scheme 7b). This result is in line with the nitrene insertion mechanism of traditional Cadogan reaction. To evaluate the electronic effect of substrates on the yield, we studied Hammett linear free energy relationship. A plot of log(kX/kH) versus constant σp of C5 functional groups attached at o-nitrobiphenyl (X=OMe, F, Cl, CF3) affords a good linear fit (ρ=1.075) in the present system. This result is in line with linear free energy relationships obtained by Cadogan and Radosevich (Scheme 7c) [12b].

|
Download:
|
| Scheme 7. Mechanistic studies. (a) Stern-Volmer quenching of 1a or/and PPh3 to 4CzIPN. (b) Kinetic isotope effect. (c) Hammett plot for carbazole formation, Equation: y=1.075x – 0.032; R2=0.9489. (d) DFT calculations for the initial deoxygenation step and proposed photoactivating models. | |
{kind=link}
Further, we did density functional theory (DFT) calculations to support the proposed photoactivation model (Scheme 7d). First, the electron-poor nitroarene 1a and the electron-rich PPh3 interact via a coulombic attraction with a P-O distance of 3.57 Å and a π−π interaction with a shortest π-stacking distance of approximately 3.44 Å. This assembly leads to the formation of the atom transfer complex Int-1. It is calculated to be endergonic by 2.2 kcal/mol, which is much lower than the generation of the dioxazaphosphetane intermediate from trialkyl phosphines including Radosevich's phosphacycloalkane [12b]. Hence, in our method, the energy transfer from the excited 4CzIPN to Int-1 promotes it to its excited state, which then undergoes oxygen transfer to give nitrosobiphenyl and P(O)Ph3. These results could explain why the photocatalysis system work at mild thermal temperature but much lower than traditional Cadogan cyclization. Moreover, the time-dependent DFT (TM-DFT) calculation (Fig. S12 in Supporting information) on the excited state of Int-1 assigned the S0-to-S2 excitation with an excitation energy of 3.52 eV. This peak was predicted to be 352 nm and mainly contributed by local excitation (92%) involving the nitrobiphenyl π orbitals (HOMO-1 to LUMO).
In summary, we have developed a mild visible-light-induced photoredox Cadogan cyclization for the synthesis of carbazoles and related heterocycles using 4CzIPN as photosensitizer. Hence, the photoacitivating action model based on energy transfer have been discovered to display unique kinetic characteristics. The mild photochemical protocol affords a scalable metal-free access to a broad range of azaheterocycles including carbazoles, benzocarbazoles, azacarbazole, azabenzocarbazoles, and indoles bearing various compatible functionalities. The versatile photoredox systems have also been demonstrated viable for intramoculelar reductive N-N couplings that enable a facile synthesis of structurally significant pyrido[1, 2-b]indazoles. These findings may inspire other reactivity discovery of mild reductive aminations.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 22071211), the Science and Technology Planning Project of Hunan Province (No. 2019RS2039), Hunan Provincial Natural Science Foundation of China (No. 2020JJ3032), and the Collaborative Innovation Center of New Chemical Technologies for Environmental Benignity and Efficient Resource Utilization.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.047.
| [1] |
(a) A.W. Schmidt, K.R. Reddy, H.J. Knolker, Chem. Rev. 112 (2012) 3193–3328; (b) T. Janosik, A. Rannug, U. Rannug, et al., Chem. Rev. 118 (2018) 9058–9128. |
| [2] |
(a) D. Gendron, M. Leclerc, Energy Environ. Sci. 4 (2011) 1225–1237; (b) Y. Ooyama, S. Inoue, T. Nagano, et al., Angew. Chem. Int. Ed. 50 (2011) 7429-7433; (c) Z. He, C. Zhang, X. Xu, et al., Adv. Mater. 23 (2011) 3086–3089; (d) M.S. Gong, J.R. Cha, C.W. Lee, Org. Electron. 42 (2017) 66–74. |
| [3] |
(a) G.R. Humphrey, J.T. Kuethe, Chem. Rev. 106 (2006) 2875–2911; (b) D.F. Taber, P.K. Tirunahari, Tetrahedron 67 (2011) 7195–7210; (c) J. Roy, A.K. Jana, D. Mal, Tetrahedron 68 (2012) 6099–6121; (d) W.C.P. Tsang, R.H. Munday, G. Brasche, et al., J. Org. Chem. 73 (2008) 7603–7610; (e) S.H. Cho, J. Yoon, S. Chang, J. Am. Chem. Soc. 133 (2011) 5996–6005; (f) H. Gao, Q.L. Xu, M. Yousufuddin, et al., Angew. Chem. Int. Ed. 53 (2014) 2701–2705; (g) J.Y. Chen, C.T. Zhong, Q.W. Gui, et al., Chin. Chem. Lett. 32 (2021) 475–479. |
| [4] |
B.P. Mundy, M.G. Ellerd, J.G. Frank Favaloro, Name Reactions and Reagents in Organic Synthesis, 2nd Ed., John Wiley & Sons, Inc., 2005.
|
| [5] |
(a) A. Kaga, S. Chiba, ACS Catal. 7 (2017) 4697–4706; (b) J.K. Matsui, S.B. Lang, D.R. Heitz, et al., ACS Catal. 7 (2017) 2563–2575; (c) H. Yi, G. Zhang, H. Wang, et al., Chem. Rev. 117 (2017) 9016–9085. |
| [6] |
(a) P. Muller, C. Fruit, Chem. Rev. 103 (2003) 2905–2919; (b) M.P. Doyle, R. Duffy, M. Ratnikov, et al., Chem. Rev. 110 (2010) 704–724; (c) D. Huang, G. Yan, Adv. Synth. Catal. 359 (2017) 1600–1619; (d) C. Wentrup, Chem. Rev. 117 (2017) 4562–4623. |
| [7] |
(a) P. Gandeepan, T. Muller, D. Zell, et al., Chem. Rev. 119 (2019) 2192–2452; (b) Y. Park, Y. Kim, S. Chang, Chem. Rev. 117 (2017) 9247–9301; (c) J. Jiao, K. Murakami, K. Itami, ACS Catal. 6 (2015) 610–633. |
| [8] |
(a) R.J. Sundberg, L.S. Lin, D.E. Blackburn, J. Heterocycl. Chem. 6 (1969) 441; (b) R.J. Sundberg, H.F. Russell, W.V. Ligon, et al., J. Org. Chem. 38 (1972) 719–724. |
| [9] |
(a) P.J. Bunyan, J.I.G. Cadogan, J. Chem. Soc. (1963) 42–49; (b) J.I.G. Cadogan, M. Cameron-Wood, R.K. Mackie, et al., J. Chem. Soc. (1965) 4831–4837; (c) J.I.G. Cadogan, Q. Rev., Chem. Soc. 22 (1968) 222–251; (d) R. Sanz, J. Escribano, M.R. Pedrosa, et al., Adv. Synth. Catal. 349 (2007) 713–718; (e) F. Martínez-Lara, A. Suárez, S. Suárez-Pantiga, et al., Org. Chem. Front. 7 (2020) 1869–1877. |
| [10] |
A.W. Freeman, M. Urvoy, M.E. Criswell, J. Org. Chem. 70 (2005) 5014-5019. DOI:10.1021/jo0503299 |
| [11] |
(a) N.E. Genung, L. Wei, G.E. Aspnes, Org. Lett. 16 (2014) 3114–3117; (b) F. Zhou, D.S. Wang, T.G. Driver, Adv. Synth. Catal. 357 (2015) 3463–3468; (c) M. Shevlin, X. Guan, T.G. Driver, ACS Catal. 7 (2017) 5518–5522; (d) H. Song, Z. Yang, C.H. Tung, W. Wang, ACS Catal. 10 (2020) 276–281. |
| [12] |
(a) T.V. Nykaza, T.S. Harrison, A. Ghosh, et al., J. Am. Chem. Soc. 139 (2017) 6839–6842; (b) T.V. Nykaza, A. Ramirez, T.S. Harrison, et al., J. Am. Chem. Soc. 140 (2018) 3103–3113; (c) G. Li, T.V. Nykaza, J.C. Cooper, et al., J. Am. Chem. Soc. 142 (2020) 6786–6799. |
| [13] |
(a) T. Bach, J.P. Hehn, Angew. Chem. Int. Ed. 50 (2011) 1000–1045; (b) D.M. Schultz, T.P. Yoon, Science 343 (2014) 1239176; (c) J. Xuan, W.J. Xiao, Angew. Chem. Int. Ed. 51 (2012) 6828–6838; (d) T.P. Yoon, M.A. Ischay, J. Du, Nat. Chem. 2 (2010) 527–532; (e) C.K. Prier, D.A. Rankic, D.W. MacMillan, Chem. Rev. 113 (2013) 5322–5363; (f) W.B. He, L.Q. Gao, X.J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1895–1898; (g) W. Yang, B. Li, M. Zhang, et al., Chin. Chem. Lett. 31 (2020) 1313–1316; (h) S. He, X. Chen, F. Zeng, et al., Chin. Chem. Lett. 31 (2020) 1863–1867; (i) G.H. Li, Q.Q. Han, Y.Y. Sun, et al., Chin. Chem. Lett. 31 (2020) 3255–3258; (j) X. Mi, Y. Kong, J. Zhang, et al., Chin. Chem. Lett. 30 (2019) 2295–2298; (k) C. Wang, X. Huang, X. Liu, et al., Chin. Chem. Lett. 31 (2020) 677–680; (l) L. Wang, M. Zhang, Y. Zhang, et al., Chin. Chem. Lett. 31 (2020) 67–70; (m) W. Xiao, J. Wu, Chin. Chem. Lett. 31 (2020) 3083–3094. |
| [14] |
(a) A.C. Hernandez-Perez, S.K. Collins, Angew. Chem. Int. Ed. 52 (2013) 12696–12700; (b) S. Choi, T. Chatterjee, W.J. Choi, et al., ACS Catal. 5 (2015) 4796–4802; (c) A.C. Hernandez-Perez, A. Caron, S.K. Collins, Chem. Eur. J. 21 (2015) 16673–16678; (d) S. Parisien-Collette, A.C. Hernandez-Perez, S.K. Collins, Org. Lett. 18 (2016) 4994–4997; (e) T. Chatterjee, G.B. Roh, M.A. Shoaib, et al., Org. Lett. 19 (2017) 1906–1909; (f) J. Shen, N. Li, Y. Yu, et al., Org. Lett. 21 (2019) 7179–7183. |
| [15] |
(a) X.D. Xia, J. Xuan, Q. Wang, et al., Adv. Synth. Catal. 356 (2014) 2807–2812; (b) S. Parisien-Collette, C. Cruché, X. Abel-Snape, et al., Green Chem. 19 (2017) 4798–4803; (c) L. Yang, Y. Zhang, X. Zou, et al., Green Chem. 20 (2018) 1362–1366. |
| [16] |
(a) C. Lu, Z. Su, D. Jing, et al., Org. Lett. 21 (2019) 1438–1443; (b) S. Peng, Y. Lin, W. He, Chin. J. Org. Chem. 40 (2020) 541; (c) Z. Wang, Q. Liu, X. Ji, et al., ACS Catal. 10 (2020) 154–159; (d) W. Ou, R. Zou, M. Han, et al., Chin. Chem. Lett. 31 (2020) 1899–1902; (e) H. Huang, K. Deng, G.J. Deng, Green Chem. 22 (2020) 8243–8247; (f) X. Ji, Q. Liu, Z. Wang, et al., Green Chem. 22 (2020) 8233–8237. |
| [17] |
(a) E. Arceo, I.D. Jurberg, A. Alvarez-Fernandez, et al., Nat. Chem. 5 (2013) 750–756; (b) S.R. Kandukuri, A. Bahamonde, I. Chatterjee, et al., Angew. Chem. Int. Ed. 54 (2015) 1485–1489; (c) L. Wozniak, J.J. Murphy, P. Melchiorre, J. Am. Chem. Soc. 137 (2015) 5678–5681; (d) A. Fawcett, J. Pradeilles, Y. Wang, et al., Science 357 (2017) 283–286; (e) J. Wu, L. He, A. Noble, et al., J. Am. Chem. Soc. 140 (2018) 10700–10704. |
| [18] |
G. Pandey, D. Pooranch, U.T. Bhalerao, Tetrahedron 47 (1991) 1745-1752. DOI:10.1016/S0040-4020(01)96916-9 |
| [19] |
J. Luo, J. Zhang, ACS Catal. 6 (2016) 873-877. DOI:10.1021/acscatal.5b02204 |