 2021, Vol. 32
2021, Vol. 32 


b Antibiotics Research and Re-evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, School of Pharmacy, Chengdu University, Chengdu 610052, China
The control of stereoselectivity is one of the central themes of current organic synthesis. Developing synthetic strategies to obtain the full set of stereoisomers of an important molecules is of great significance [1]. For example, stereoisomers of a drug molecule may exhibit different biological activities. Stereodivergent catalysis is an ideal strategy, which can achieve precise control of the relative configuration and absolute configuration by changing the reaction conditions, thereby allow efficient access to multiple stereoisomers of a given product from the same set of starting materials [2]. Using different chiral catalysts [3], central metals [4], ligands [5], additives [6], or utilizing synergistic catalytic systems are common strategies to achieve stereodivergent catalysis [7]. So far, there are very few reports on controlling the stereoselectivity by changing the reaction medium, and it is generally considered difficult to reverse the inherently preferred diastereoselectivity by simply changing the reaction solvent [8].
Spirooxindole is a core structure of many natural products and bioactive molecules. Therefore, it has aroused great interest of researchers from academic and industrial fields [9]. Despite the enormous progress on the construction of chiral spirooxindoles, the stereodivergent synthesis of spirooxindoles containing multiple stereocenters remains a significant challenge and there are only a handful of strategies were reported [10]. In 2007, Trost et al. described a palladium-catalyzed diastereodivergent [3+2] trimethylenemethane cycloaddition, successfully producing cis- or trans-spirocyclic oxindolic cyclopentanes by changing the ligands (Fig. 1a) [10a]. In 2016, Chen and co-workers achieved the diastereodivergent synthesis of bis(spirocyclic) oxindoles through [3+2] annulations catalysed by different types of Lewis bases (Fig. 1b) [10b]. Considering the significance of the privileged spirooxindole scaffolds, exploitation of new strategy for the stereodivergent synthesis of these compounds is still highly desirable.

|
Download:
|
| Fig. 1. Stereodivergent synthesis of spirocyclopentaneoxindoles. | |
Retro-aldol reaction is a useful method for cleaving the carbon−carbon bond and its synthetic utility has been demonstrated in total synthesis, kinetic resolution, and various cascade processes [11]. However, in contrast to extensive studies on the catalytic aldol reaction, the catalytic retro-aldol reaction has been much less explored. So far, antibodies, amines, inorganic bases, and several metals have been reported to promote this type of reaction [12]. N-Heterocyclic carbene (NHC), which can function either as a Brønsted base catalyst or Lewis base catalyst, may serve as an efficient catalyst for the retro-aldol reaction and provide new opportunities for stereodivergent synthesis through cascade process [13].
We report here a novel sequential process for the stereodivergent synthesis of spirocyclopentaneoxindole derivatives (Fig. 1c). At the first stage, intermediate I was assembled through Michael/aldol reaction catalysed by a chiral secondary amine. Notably, at the second stage, a retro-aldol/aldol cascade reaction was achieved via NHC catalysis, affording chiral spirocyclopentaneoxindole products with four consecutive stereocenters. Intriguingly, this ring opening-closure process enables the divergent synthesis of two diastereoisomers by simply changing the solvents. Using different combinations of (R)- or (S)-chiral amine and two different solvents, four stereoisomers can be selectively provided.
We initiated our study by investigating the reaction of N-benzyl-3-(2-oxo-2-phenylethyl)oxindole 1a with 4-chlorocinnam-aldehyde 2a in the presence of chiral secondary amine catalysts C1-C5 with acetic acid as an additive. Michael/aldol product 3a was obtained as the major product through the iminium/enamine cascade process without dehydration [14]. To our delight, TMS-protected diphenylprolinol C2 proves to be the optimum catalyst. Further screening of other reaction parameters (Table S1 in Supporting information) uncovered toluene and acetic acid as the optimum solvent and additive for the generation of 3a in 85% yield with > 19:1 d.r. and 99:1 e.r. (Table 1, entry 2). Changing C2 to ent-C2, the enantiomer of product 3a was generated successfully in 87% yield with > 19:1 d.r. and 6:94 e.r. (Table 1, entry 6).
|
|
Table 1 Evaluation of conditions for the asymmetric Michael/aldol reaction. |
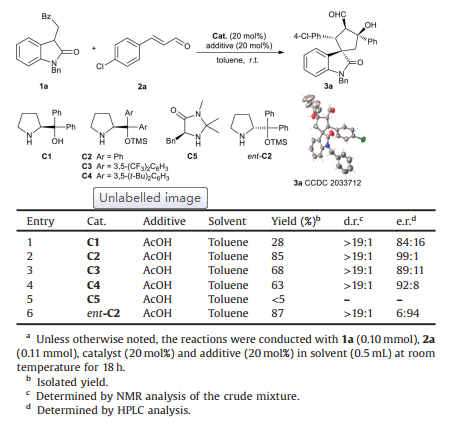
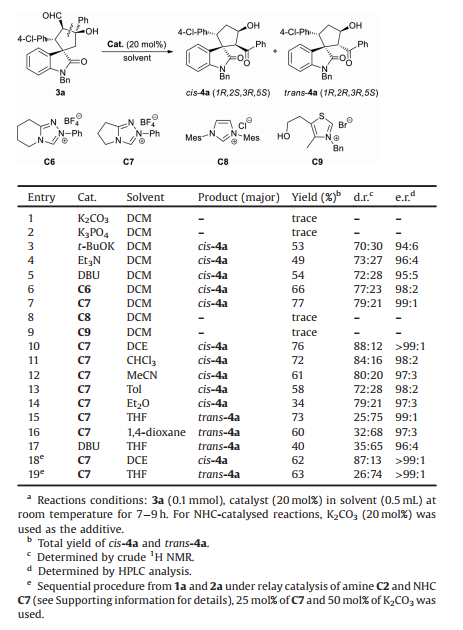
After established the optimum conditions for the Michael/aldol reaction, we next explored the intramolecular retro-aldol/aldol cascade reaction of the enantioenriched β-hydroxyaldehyde 3. To promote the C-C bond cleavage, we first screened a series of bases. As shown in Table 2, the reaction proceeded very slowly in the presence of K2CO3 or K3PO4 in dichloromethane at room emperature (Table 2, entries 1 and 2). Then, potassium tert-butoxide (t-BuOK), 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU), and triethylamine (Et3N) were tested (Table 2, entries 3–5). Products cis-4a and trans-4a, which are epimers containing four consecutive stereocenters, could be obtained in moderate yields (49%–54%) with moderate diastereoselectivity (70:30 to 73:27 d.r.) and slightly declined e.r. values. Next, we screened a series of NHCs as the catalyst using K2CO3 as the additive (Table 2, entries 6–9). Interestingly, triazolium C6 led to the formation of cis-4a as the major product in 66% yield with better diastereoselectivity (77:23 d.r., Table 2, entry 6).
|
|
Table 2 Effects of catalyst and solvent on the retro-aldol/aldol cascade reaction. |
{kind=link}
To our delight, less steric hindered C7 could further improve the yield and diastereoselectivity (77% yield, 79:21 d.r., Table 2, entry 7). The type of NHC catalyst is important for the retro aldol/aldol reaction. Imidazole-based C8 and thiazole-based C9 showed very low catalytic activity in this transformation (Table 2, entries 8 and 9). Subsequently, we evaluated the reactions in different solvents. Using DCE as the solvent led to the best diastereoselectivity (88:12 d.r.) without significantly affecting the yield and enantioselectivity (Table 2, entry 10). Cis-4a was also generated as the major product in chloroform, acetonitrile, toluene, or ethyl ether (Table 2, entries 11–14). Intriguingly, in sharp contrast with the formation of cis-4a as the major product in the aforementioned reaction media, the reaction in tetrahydrofuran (THF) afforded trans-4a as the major isomer in 73% yield with 75:25 d.r. and 99:1 e.r. (Table 2, entry 15). The reaction using 1, 4-dioxane as the solvent also provided trans-4a as the major product, albeit with declined yield and diastereoselectivity (Table 2, entry 16). DBU catalysed reaction in THF only gave trans-4a in low yield with moderate diastereoselectivity (Table 2, entry 17). To establish a convenient process for the stereodivergent construction of spirocyclopentaneoxindoles, after uncovered the key role of the NHC catalyst and the solvent-controlled switchable diastereo-selectivity, we further investigated the sequential reaction process under relay catalysis of the secondary amine and the NHC (Table 2, entries 18 and 19). Upon completion of the Michael/aldol reaction, the solvent was switched to DCE or THF, and triazolium C7 was added accompanied with K2CO3. The presence of secondary amine C2 and acetic acid had little effect on the performance of the NHC in the following retro-aldol/aldol reaction, which provided corresponding cis-4a in 54% yield with 87:13 d.r. and > 99:1 e.r. (Table 2, entry 18) or trans-4a in 47% yield with 74:26 d.r. and > 99:1 e.r. (Table 2, entry 19).
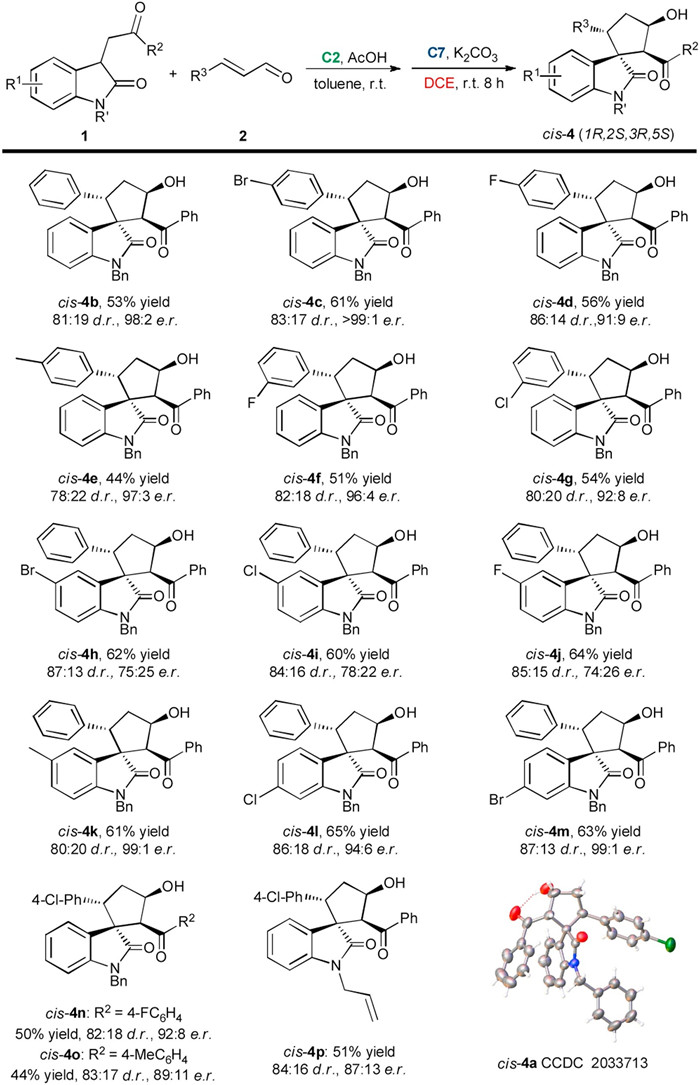
Under the optimized sequential reaction conditions, we investigated the substrate scope of this solvent-controlled stereodivergent strategy. Scheme 1, Scheme 2 summarizes the synthesis of cis- and trans-spirocyclopentaneoxindoles 4 from various substituted oxindoles 1 with different cinnamaldehydes 2 through secondary amine/NHC relay catalysis. For the cis-selective synthesis, the sequential reaction involving cinnamaldehyde 2 with various R3 groups proceeded smoothly, providing the corresponding products cis-4 in yields of 44%–61% with 78:22 to 87:13 d.r. and good to excellent enantioselectivities (cis-4a-4g). Different substituents such as halogen atoms and a methyl group at the ortho- or meta-position on the phenyl ring of cinnamaldehyde 2 were well tolerated. The relative and absolute configurations of cis-4a have been confirmed via X-ray crystallographic analysis. 3-Substituted oxindoles 1 bearing halogen atoms at the 5- or 6-position of the oxindoles showed good reactivity, affording spirocyclopentaneoxindole cis-4h-4m in 60%–65% yields with good diastereoselectivity, while halogen substituents at the 5-position led to a drop of e.r. value. Substrates 1 bearing different aryl R2 groups also smoothly reacted with 2a, producing cis-4n and cis-4o in slightly declined yields with good diastereoselectivity and enantioselectivity. However, when R2 were alkyl groups, the desired product could not be formed.

|
Download:
|
| Scheme 1. Synthesis of the cis-spirocyclopentaneoxindoles. For reactions conditions, see entry 18 in Table 2. Isolated yield of the major product and the d.r. value was determined by 1H NMR analysis of the crude reaction mixture, the e.r. value was determined by HPLC analysis. | |
{kind=link}

|
Download:
|
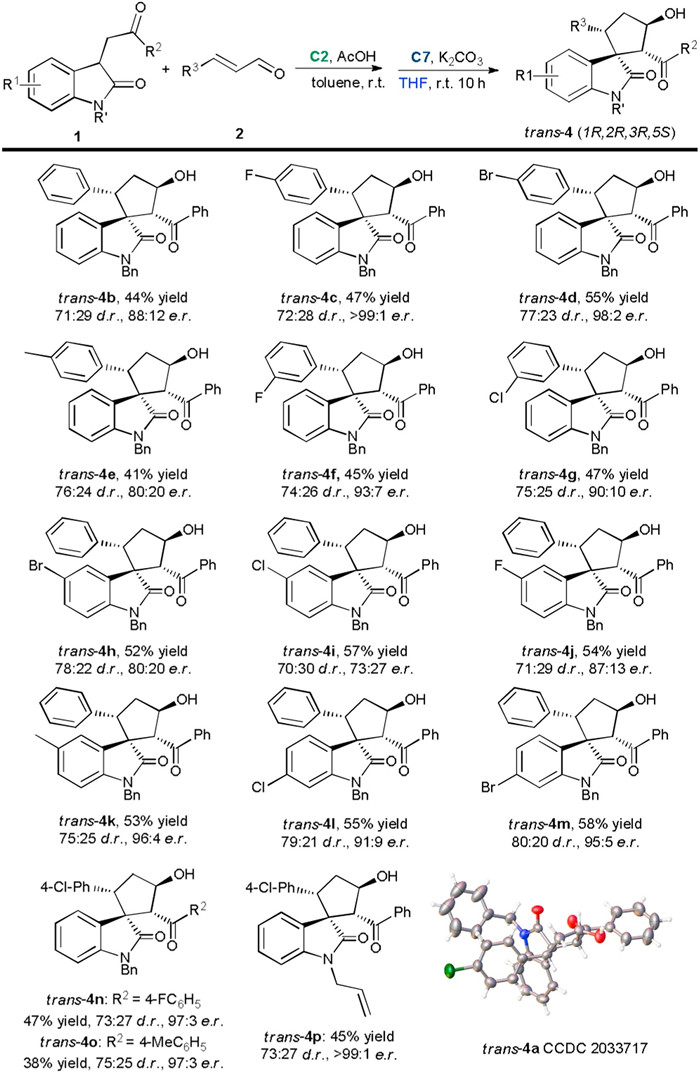
| Scheme 2. Synthesis of the trans-spirocyclopentaneoxindoles. For reactions conditions, see entry 19 in Table 2. Isolated yield of the major product; the d.r. value was determined by 1H NMR analysis of the crude reaction mixture; the e.r. value was determined by HPLC analysis. | |
{kind=link}
Changing the N-substituent of oxindole 1 to allyl group did not affect the reactivity and stereoselectivity (cis-4p). For the synthesis of trans-spirocyclopentaneoxindole, to our delight, the diastereoselectivity was successfully switched in all cases by simply changing the solvent of the second stage to THF (Scheme 2). Reactions of cinnamaldehyde 2 bearing various substituents (R3) with 3-substituted oxindoles 1 bearing different substituents (R1, R2 and R') proceeded smoothly, affording target products trans-4a-4p in acceptable yields with up to > 99:1 e.r. The relative and absolute configurations of trans-selective products were confirmed by X-ray crystallographic analysis of trans-4a.
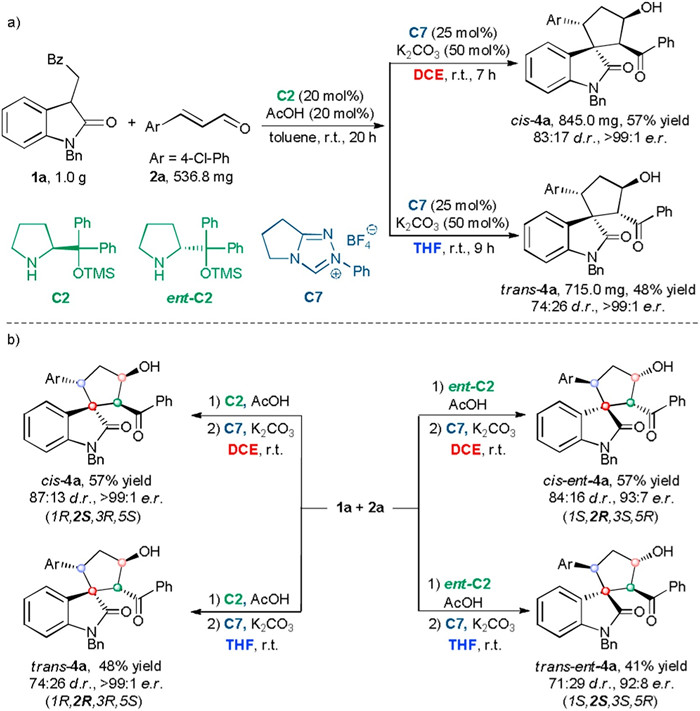
To demonstrate the synthetic usefulness of this protocol, the solvent-controlled stereodivergent sequential reactions were performed on a gram-scale (Scheme 3a). Products cis-4a and trans-4a were delivered in 57% yield and 48% yield, respectively, without affecting the stereoselectivities. Changing chiral secondary amine catalyst C2 to ent-C2 enables the stereodivergent synthesis of four stereoisomers (Scheme 3b). Cis- ent-4a was obtained in 57% yield with 84:16 d.r. and 93:7 e.r. by involving ent-C2 in the Michael/aldol reaction and DCE as the solvent in the NHC-catalyzed retro-aldol/aldol reaction. Using the combination of ent-C2 in the Michael/aldol reaction and THF in the retro-aldol/aldol reaction led to trans- ent-4a with a similar result (41% yield, 71:29 d.r., 92:8 e.r.). Thus, changing one chiral catalyst allows the assembly of four stereoisomers through the sequential process.

|
Download:
|
| Scheme 3. Gram-scale reactions and synthesis of four stereoisomers of 4a. | |
{kind=link}
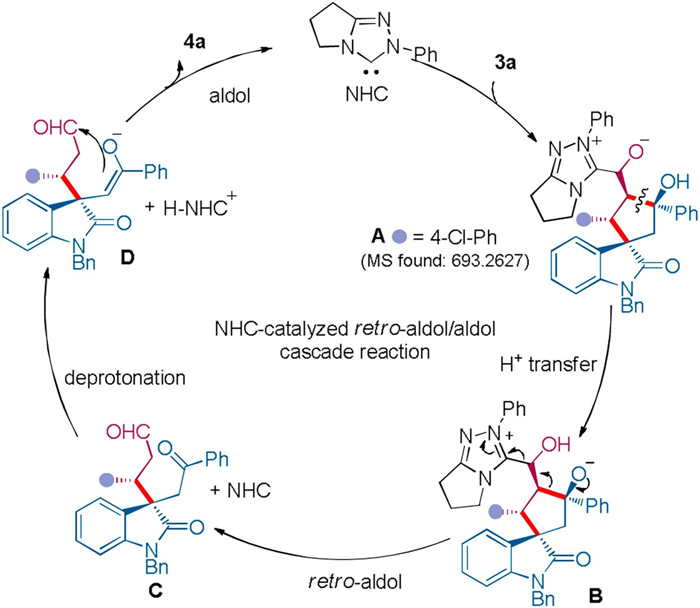
Based on the experimental results, we proposed a possible mechanism for the novel NHC-catalysed retro-aldol/aldol cascade reaction (Fig. 2). The initial addition of the NHC to β-hydroxyaldehyde 3, which was produced by the secondary amine-catalysed Michael/aldol reaction, would form intermediate A. The following intramolecular proton transfer would give intermediate B. Then, the C-C bond cleavage might generate C accompanied with the release of the NHC. Next, the triazole carbene acted as a Brønsted base to deprotonate the α-H of the ketone moiety, which led to the formation of intermediate D. Intramolecular aldol reaction of D would afford product 4 and regenerate the NHC. The mass peak of [C43H37ClN4O3+H]+ was detected in HRMS analysis of the reaction mixture, indicating the possible generation of intermediate A or B. The solvent effect on the stereochemical outcome might be rationalized by a reversal of the enantiofacial bias of the last aldol reaction, since using THF or DCE as the solvent might lead different solvation effect on the ion pair intermediates D.

|
Download:
|
| Fig. 2. Possible mechanism for the NHC-catalysed retro-aldol/aldol cascade reaction. | |
{kind=link}
In this work, we have developed a NHC-catalysed retro-aldol/aldol cascade reaction, which enables the solvent-controlled diastereodivergent synthesis of spirocyclopentane-oxindole derivatives. In the ring opening-closure process, two diastereoisomers can be obtained by simply switching the reaction solvents (THF or DCE). Moreover, the Michael/aldol/ retro-aldol/aldol sequential reaction process allows the stereodivergent synthesis of spirocyclopentane oxindoles containing four consecutive stereocenters from 3-substituted oxindole and α, β-unsaturated aldehyde through the relay catalysis of the secondary amine and NHC. Four stereoisomers of the product can be selectively provided by using different combinations of a chiral secondary amine (C2 or ent-C2) and a solvent (THF or DCE). Studies on the origin of the solvent-controlled divergent diastereoselectiviety in this cascade reaction are currently in progress in our laboratory.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe are grateful for financial support from Ministry of Science and Technology of China (No. 2019ZX09721001-008), National Natural Science Foundation of China (Nos. 81773890, 22001024, 82073997), the "Thousand Talents Program" of Sichuan Province and Xinglin Scholar Research Premotion Project of CDUTCM.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.03.009.
| [1] |
(a) G. Zhan, W. Du, Y.C. Chen, Chem. Soc. Rev. 46 (2017) 1675-1692; (b) M. Bihani, J.C.G. Zhao, Adv. Synth. Catal. 359 (2017) 534-575; (c) L. Lin, X. Feng, Chem. Eur. J. 23 (2017) 6464-6482; (d) S. Krautwald, E.M. Carreira, J. Am. Chem. Soc. 139 (2017) 5627-5639; (e) I.P. Beletskaya, C. Nájera, M. Yus, Chem. Rev. 118 (2018) 5080-5200; (f) J. Adrio, J.C. Carretero, Chem. Commun. 55 (2019) 11979-11991; (g) S.L. Rössler, D.A. Petrone, E.M. Carreira, Acc. Chem. Res. 52 (2019) 2657-2672; (h) C.Nájera, F. Foubelo, J.M. Sansano, M.Yus, Org. Biomol. Chem.18 (2020) 1232-1278; (i) L.C. Miller, R. Sarpong, Chem. Soc. Rev. 40 (2011) 4550-4562. |
| [2] |
(a) J. Mahatthananchai, A.M. Dumas, J.W. Bode, Angew. Chem. Int. Ed. 51 (2012) 10954-10990; (b) M.T. Oliveira, M. Luparia, D. Audisio, N. Maulide, Angew. Chem. Int. Ed. 52 (2013) 13149-13152. |
| [3] |
(a) X. Li, W. Huang, Y.Q. Liu, et al., J. Org. Chem. 82 (2017) 397-406; (b) S.B.J. Kan, H. Maruyama, M. Akakura, T. Kano, K. Maruoka, Angew. Chem. Int. Ed. 56 (2017) 9487-9491; (c) C. Verrier, P. Melchiorre, Chem. Sci. 6 (2015) 4242-4246; (d) X. Feng, Z. Zhou, R. Zhou, et al., J. Am. Chem. Soc. 134 (2012) 19942-19947; (e) L. Zhang, H. Yuan, W. Lin, et al., Org. Lett. 20 (2018) 4970-4974; (f) W. Xiao, Q.Q. Yang, Z. Chen, et al., Org. Lett. 20 (2018) 236-239; (g) H. Huang, S. Konda, J.C.G. Zhao, Angew. Chem. Int. Ed. 55 (2016) 2213-2216; (h) X. Li, M. Lu, Y. Dong, et al., Nat. Commun. 5 (2014) 4479; (i) S. Meninno, A. Roselli, A. Capobianco, J. Overgaard, A. Lattanzi, Org. Lett. 19 (2017) 5030-5033; (j) D. Uraguchi, K. Yoshioka, T. Ooi, Nat. Commun. 8 (2017) 14793. |
| [4] |
(a) X. Cong, G. Zhan, Z. Mo, M. Nishiura, Z. Hou, J. Am. Chem. Soc. 142 (2020) 5531-5537; (b) H.L. Teng, Y. Luo, M. Nishiura, Z. Hou, J. Am. Chem. Soc. 139 (2017) 16506-16509; (c) Y. Kuang, B. Shen, L. Dai, et al., Chem. Sci. 9 (2018) 688-692; (d) G. Lu, T. Yoshino, H. Morimoto, et al., Angew. Chem. Int. Ed. 50 (2011) 4382-4385. |
| [5] |
(a) X.X. Yan, Q. Peng, Q. Li, et al., J. Am. Chem. Soc. 130 (2008) 14362-14363; (b) M. Luparia, M.T. Oliveira, D. Audisio, et al., Angew. Chem. Int. Ed. 50 (2011) 12631-12635; (c) D. Audisio, M. Luparia, M.T. Oliveira, D. Klütt, N. Maulide, Angew. Chem. Int. Ed. 51 (2012) 7314-7317; (d) E.E. Maroto, S. Filippone, M. Suárez, et al., J. Am. Chem. Soc. 136 (2014) 705-712; (e) X. Hao, L. Lin, F. Tan, et al., ACS Catal. 5 (2015) 6052-6056; (f) M. Mechler, R. Peters, Angew. Chem. Int. Ed. 54 (2015) 10303-10307; (g) K. Weidner, Z. Sun, N. Kumagai, M. Shibasaki, Angew. Chem. Int. Ed. 54 (2015) 6236-6240; (h) C.Y. Li, W.L. Yang, X. Luo, W.P. Deng, Chem. Eur. J. 21 (2015) 19048-19057; (i) T.T. Gao, W.W. Zhang, X. Sun, H.X. Lu, B.J. Li, J. Am. Chem. Soc. 141 (2019) 4670-4677; (j) M.M. Zhang, Y.N. Wang, B.C. Wang, et al., Nat. Commun. 10 (2019) 2716; (k) X.J. Liu, S. Jin, W.Y. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 2039-2043. |
| [6] |
(a) J. Gao, S. Bai, Q. Gao, Y. Liu, Q. Yang, Chem. Commun. 47 (2011) 6716-6718; (b) Á. Martínez-Castañeda, H. Rodríguez-Solla, C. Concellón, V. del Amo, J. Org. Chem. 77 (2012) 10375-10381. |
| [7] |
(a) C.S. Schindler, E.N. Jacobsen, Science 340 (2013) 1052-1053; (b) B. Han, X. Xie, W. Huang, et al., Adv. Synth. Catal. 356 (2014) 3676-3682; (c) N.K. Rana, H. Huang, J.C.G. Zhao, Angew. Chem. Int. Ed. 53 (2014) 7619-7623; (d) L. Næsborg, K.S. Halskov, F. Tur, S.M.N. Mønsted, K.A. Jørgensen, Angew. Chem. Int. Ed. 54 (2015) 10193-10197; (e) T. Sandmeier, S. Krautwald, H.F. Zipfel, E.M. Carreira, Angew. Chem. Int. Ed. 54 (2015) 14363-14367; (f) P. Chen, Y. Li, Z.C. Chen, W. Du, Y.C. Chen, Angew. Chem. Int. Ed. 59 (2020) 7083-7088; (g) X. Huo, R. He, X. Zhang, W. Zhang, J. Am. Chem. Soc. 138 (2016) 11093-11096; (h) Z.T. He, X. Jiang, J.F. Hartwig, J. Am. Chem. Soc. 141 (2019) 13066-13073; (i) R. He, X. Huo, L. Zhao, et al., J. Am. Chem. Soc. 142 (2020) 8097-8103; (j) L. Wei, Q. Zhu, S.M. Xu, X. Chang, C.J. Wang, J. Am. Chem. Soc. 140 (2018) 1508-1513; (k) S. Krautwald, M.A. Schafroth, D. Sarlah, E.M. Carreira, J. Am. Chem. Soc. 136 (2014) 3020-3023; (l) S. Krautwald, D. Sarlah, M.A. Schafroth, E.M. Carreira, Science 340 (2013) 1065-1068. |
| [8] |
(a) M. Morgen, S. Bretzke, P. Li, D. Menche, Org. Lett. 12 (2010) 4494-4497; (b) A. Katsuyama, A. Matsuda, S. Ichikawa, Org. Lett. 18 (2016) 2552-2555; (c) X. Tian, C. Cassani, Y. Liu, et al., J. Am. Chem. Soc. 133 (2011) 17934-17941; (d) A. Katsuyama, A. Paudel, S. Panthee, et al., Org. Lett. 19 (2017) 3771-3774; (e) A. Katsuyama, F. Yakushiji, S. Ichikawa, J. Org. Chem. 83 (2018) 7085-7101; (f) P.V. Ramachandran, P.B. Chanda, Org. Lett. 14 (2012) 4346-4349. |
| [9] |
(a) Z.Y. Cao, J. Zhou, Org. Chem. Front. 2 (2015) 849-858; (b) Y. Wang, H. Lu, P.F. Xu, Acc. Chem. Res. 48 (2015) 1832-1844; (c) Z.Y. Cao, F. Zhou, J. Zhou, Acc. Chem. Res. 51 (2018) 1443-1454; (d) Q. Zhao, C. Peng, H. Huang, et al., Chem. Commun. 54 (2018) 8359-8362; (e) G.J. Mei, F. Shi, Chem. Commun. 54 (2018) 6607-6621; (f) K.K. Wang, W. Du, J. Zhu, Y.C. Chen, Chin. Chem. Lett. 28 (2017) 512-516; (g) N.Y. Chen, L.P. Ren, M.M. Zou, et al., Chin. Chem. Lett. 25 (2014) 197-200. |
| [10] |
(a) B.M. Trost, N. Cramer, S.M. Silverman, J. Am. Chem. Soc. 129 (2007) 12396-12397; (b) G. Zhan, M.L. Shi, Q. He, et al., Angew. Chem. Int. Ed. 55 (2016) 2147-2151; (c) B. Wang, X.H. Wang, W. Huang, et al., J. Org. Chem. 84 (2019) 10349-10361; (d) X. Zhao, J. Xiong, J. An, et al., Org. Chem. Front. 6 (2019) 1989-1995; (e) B. Wang, F. Peng, W. Huang, et al., Acta Pharm. Sin. B 10 (2020) 1492-1510; (f) P.D. Chaudhari, B.C. Hong, G.H. Lee, Org. Lett. 19 (2017) 6112-6115; (g) Y. He, Y. Liu, Y. Liu, et al., Adv. Synth. Catal. 362 (2020) 2052-2058. |
| [11] |
(a) G. Zhong, D. Shabat, B. List, et al., Angew. Chem. Int. Ed. 37 (1998) 2481-2484; (b) S. Beltrán-Rodil, J.R. Donald, M.G. Edwards, et al., Tetrahedron Lett. 50 (2009) 3378-3380; (c) M.E. Jung, J.J. Chang, Org. Lett. 14 (2012) 4898-4901; (d) Y. Yan, L. Feng, G. Li, et al., ACS Catal. 7 (2017) 4473-4478; (e) H. Nonaka, Y. Nakanishi, S. Kuno, et al., Nat. Commun. 10 (2019) 876. |
| [12] |
(a) M.E. Flanagan, J.R. Jacobsen, E. Sweet, P.G. Schultz, J. Am. Chem. Soc. 118 (1996) 6078-6079; (b) P.R. Carlier, C.W.S. Lo, M.M.C. Lo, N.C. Wan, I.D. Williams, Org. Lett. 2 (2000) 2443-2445; (c) S. Cai, Z. Liu, W. Zhang, X. Zhao, D.Z. Wang, Angew. Chem. Int. Ed. 50 (2011) 11133-11137; (d) C. Richter, M. Krumrey, M. Bahri, S. Trunschke, R. Mahrwald, ACS Catal. 6 (2016) 5549-5552; (e) S.L. Zhang, Z.Q. Deng, Org. Biomol. Chem. 14 (2016) 8966-8970; (f) S.L. Zhang, Z.L. Yu, Org. Biomol. Chem. 14 (2016) 10511-10515; (g) R. Maity, C. Gharui, A.K. Sil, S.C. Pan, Org. Lett. 19 (2017) 662-665; (h) S. Liu, T. Zhang, L. Zhu, et al., Chem. Commun. 54 (2018) 13551-13554; (i) J. Wang, Z.X. Deng, C.M. Wang, et al., Org. Lett. 20 (2018) 7535-7538; (j) F. Liu, L. Zhu, T. Zhang, et al., ACS Catal. 10 (2019) 1256-1263; (k) Y.X. Song, D.M. Du, Adv. Synth. Catal. 361 (2019) 5042-5049; (l) R.A.S. Pike, R.R. Sapkota, B. Shrestha, et al., Org. Lett. 22 (2020) 3268-3272. |
| [13] |
(a) E. Sánchez-Larios, J.M. Holmes, C.L. Daschner, M. Gravel, Org. Lett. 12 (2010) 5772-5775; (b) X. Yang, P.K. Majhi, H. Chai, et al., Angew. Chem. Int. Ed. 60 (2021) 159-165; (c) J. Kaeobamrung, J.W. Bode, Org. Lett. 11 (2009) 677-680. |
| [14] |
K. Albertshofer, K.E. Anderson, C.F. Barbas Ⅲ, Org. Lett 14 (2012) 5968-5971. DOI:10.1021/ol302876c |