 2021, Vol. 32
2021, Vol. 32 


As a group of biological active molecules, porphyrins and their derivatives have received considerable attention of chemists for a long time [1]. These molecules have been widely used in dye-sensitized solar cells (DSC) [2], nonlinear optical materials [3] and dyes for photodynamic therapy (PDT) [4] due to their distinctive electrochemical and photochemical properties. To modulate these properties, numerous scientists endeavored in the decoration of porphyrins, the most fundamental way of which is to fix functional groups to the meso or/and β position of the porphyrin core.
Functional groups containing sulfur element usually affect the main body of a molecule significantly. Porphyrins bearing 2-pyridylthio groups in meso positions were showed to be oxidized readily to form cationic species [5]. Crossley demonstrated that the introduction of PhS- to the β position of tetraphenyl porphyrinato Cu(Ⅱ) had a strong influence on its redox potentials and maximum absorption wavelengths in UV–vis spectrum [6]. This phenomenon could be partly rationalized by the strong interaction between phenylthio groups and a1u-like orbital of porphyrin unit. Scheiner pointed out that phenylthio/mercapto can lead to an inversion of a1u-like and a2u-like orbital ordering in energy [7].
Various methods have been explored to achieve porphyrins involving phenylthio groups regardless of the position attached [8-11]. These methods can be generally classified into three groups: 1) Prefunctionalization of pyrrole and sequential condensation-oxidation [9], which has succeeded in giving a series of porphyrin-tetrathiafulvalene (TTF) hybrids [9a-d], 2) late transition-metal-catalyzed C–S bond formation [10] and 3) classical SNAr reaction [11]. Despite of these powerful tools in hand, systematic study on multi-phenylthio substituted porphyrin is still limited as far as we know.
Confronting the wide application and the strong yet subtle influence of phenylthio groups on the frontier molecular orbitals (FMO) of porphyrin core, we devote to synthesize a series of multi-(phenylthio)porphyrins. Comparisons on their electro-/photochemical properties will contribute to a better understanding of the importance of these phenylthio groups' amounts and positions. The intramolecular cooperative effects of these phenylthio groups will be elucidated as well.
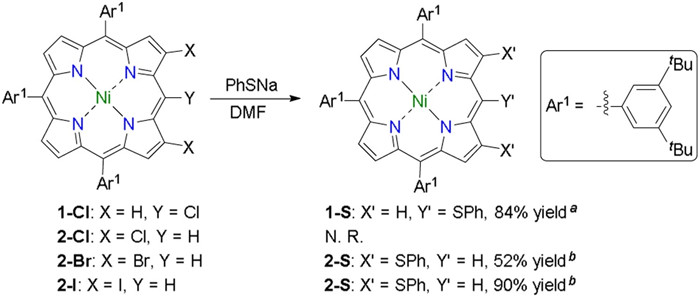
Classical SNAr reaction was selected as an ideal strategy in our syntheses due to its high atom economy and universality. Based on this strategy reactions between PhSNa and halogenated porphyrin were examined. 5, 10, 15-Tris(3, 5-di- tert-butylphenyl)-20-(phenylthio)porphyrinato Ni(Ⅱ) (1-S) could be obtained from 1-Cl smoothly at room temperature (Scheme 1). Bromo or iodo analogues of 1-Cl could afford 1-S in a similar yield. 5, 10, 15-Tris(3, 5-di- tert-butylphenyl)-2, 18-bis(phenylthio)porphyrinato Ni(Ⅱ) (2-S) could generate from 2-I under a similar condition with a yield of 90%. It was worth noting that 2-Br, the bromo analogue of 2-I, gave 2-S with a yield of 52% under a similar condition while 2-Cl did not react with PhSNa at all.

|
Download:
|
| Scheme 1. Model reactions between halogenated porphyrins and PhSNa for the syntheses of meso- and β-(phenylthio)porphyrins. Conditions: (a) PhSNa (5 equiv.), DMF, r.t., overnight; (b) PhSNa (10 equiv.), DMF, 110 ℃, overnight. | |
Based on the results of the model reactions above, we designed a series of multi-halogenated porphyrins as precursors for the achieving of corresponding multi-phenylthio porphyrins via SNAr reactions (Table 1). Multi-halogenated porphyrins 3 and 4 were synthesized according to the strategy of Yorimitsu and Osuka [12-14]. 3-S and 4-S could be given thereafter in 72% and 64% yield, respectively. These relatively low yields comparing with that of 2-S could be ascribed to the spatial repulsion between phenylthio groups. Contrast to multi-halogenated triaryl porphyrin, multi-halogenated diaryl porphyrins suffered a poor solubity, and therefore 6-S required an elevated temperature of 140 ℃ to be transformed from 6. As the Ar1 analogue of 9, 9′ showed much poorer solubility than that of 6, and thus either chlorination or SNAr reaction of 9′ was not feasible. Therefore an aryl bearing two hexyls, Ar2, was designed and introduced into this system. Owning to these flexible hexyls 10 could be obtained through chlorination of 9. Since both 9 and 10 were soluble in DMF the following SNAr reactions gave 9-S and 10-S in moderate yields.
|
|
Table 1 Syntheses of multi-(phenylthio)porphyrins. |

All of these products were well characterized by 1H NMR, 13C NMR and HRMS. Structures of 2-S, 4-S and 10-S were further confirmed by single crystal X-ray diffraction (Fig. 1). The solid state structures indicate that the porphyrin core of 3-S is almost planar with a mean-plane deviation of 0.066(2) Å. As a contrast the porphyrin core of 4-S illustrates a ruffled conformation, which can be attributed to the repulsion of three spatial adjacent phenylthio groups. However, the conformation of hexaphenylthio substituted 10-S differs from that of 4-S, this can be rationalized by the unbalanced torsion originated from peripheral six phenylthio groups. Aside from the conformation of porphyrin core, the orientations of Ph are also highly affected by the amount of substituent groups. In the molecule of 2-S, both Ph-S bonds are almost perpendicular to the porphyrin core. However, in either 4-S or 10-S Ph-S bonds attached to β positions are nearly parallel to the porphyrin plane whilst Ph-S bonds attached to meso positions are almost perpendicular to the porphyrin plane.

|
Download:
|
| Fig. 1. X-ray crystal structures of 2-S, 4-S and 10-S. (a, c, e) Top view of 2-S, 4-S and 10-S; (b, d, f) Side view of 2-S, 4-S and 10-S. Hexyl, tert-butyl, hydrogen atoms and solvents are omitted for clarity. | |
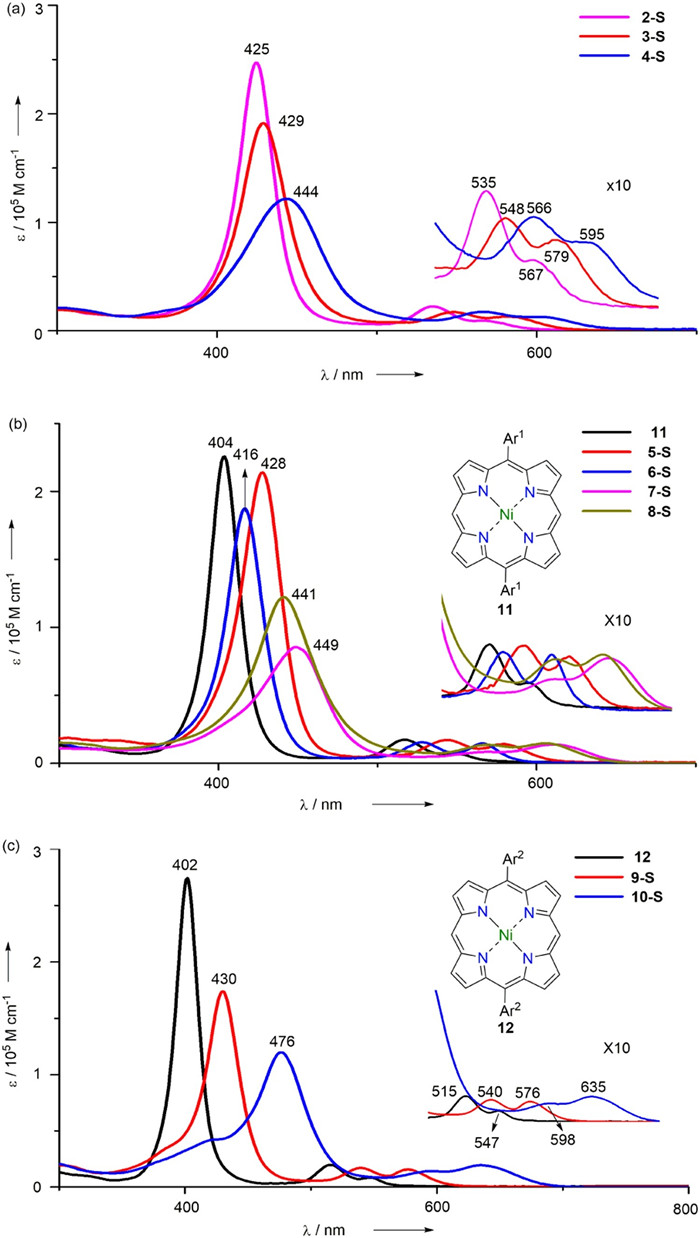
Previous research [6] indicated that S-containing substituents at β position have strong influences on the UV–vis absorption spectra of porphyrins. Hence the UV–vis spectra of these multi-(phenylthio)porphyrins were recorded to elucidate the effect of substituents' position, amount and mutual interactions. As illustrated in Fig. 2a, both 3-S and 4-S exibit red-shifted and broadened absorption bands comparing with 2-S, while 4-S shows more red-shifted and broadened peaks for both Soret and Q bands. Although red-shifted and broadened absorption in UV–vis spectra are often ascribed to the aggregation, aggregation is excluded here through comparison of a series of spectra of 3-S and 4-S with various concertration. Thus these differences can be mainly ascribed to their higher degree of substitution. In Fig. 2b the UV–vis spectrum of 5-S illustrates a Soret peak at 428 nm and two Q peaks at 545 and 580 nm. However, the β-analogue 6-S gives a less red-shifted spectrum comparing with that of 5-S, indicating the remarkable influence of phenylthio groups' position. Both 7-S and 8-S show much red shifted Soret peaks and Q peaks comparing with either 5-S or 6-S, unambiguously revealing the bathochromic effect of the increasing amount of phenylthio groups. However the differences between the UV–vis spectrum of 7-S and that of 8-S are difficult to interpret by simply considering the amount and positions of phenylthio groups involved in these molecules. Therefore TD-DFT calculation was performed and the results indicate that the significant transitions of 8-S is much more widely distributed in Soret region than those of 7-S, which leads to a smaller extinction coefficient and broader Soret peak of 8-S. As shown in Fig. 2c, 10-S demonstrates one Soret band at 476 nm and two ill-defined peaks at 598 and 635 nm, respectively. Despite of its most bathochromic shifted wavelength among these compounds, it is quite reasonable since it bears six phenylthio units.

|
Download:
|
| Fig. 2. UV–vis absorption spectra of multi-(phenylthio)porphyrins and related compounds in CH2Cl2. | |
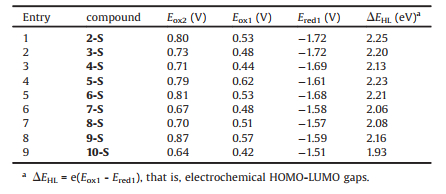
To investigate the redox properties of multi-(phenylthio)porphyrins, cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were applied. Each of these compounds demonstrates two reversible oxidation peaks and one less reversible reduction peak within the electrochemical window. Generally the oxidation potential decreases as the substituent degree of phenylthio groups increases (Table 2, entries 1, 2, 8, 9), proving the electron donating feature of phenylthio groups. Basically the electrochemical HOMO-LUMO gaps of these compounds are consistent with those derived from the maximum absorption wavelengths in UV–vis spectra.
|
|
Table 2 Redox potentials and electrochemical HOMO-LUMO gaps of multi-(phenylthio)porphyrins. |
{kind=link}
{kind=link}
{kind=link}
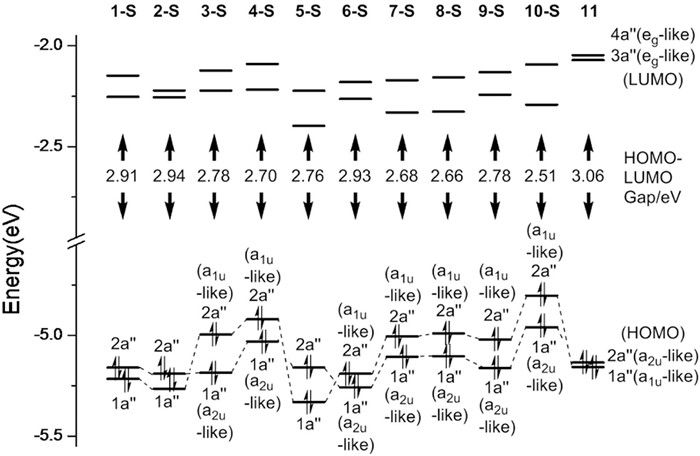
As the PhS/HS group showed strong influences on both a1u-like and a2u-like orbitals of porphyrin [6, 7], the interactions between porphyrin core and phenylthio groups within a sole molecule aroused our interest. DFT calculation reveals that the substitution of phenylthio groups leads to a decrease of HOMO-LUMO gap comparing with that of 11, while multi-(phenylthio)porphyrins showed remarkably narrower HOMO-LUMO gaps than mono(phenylthio)porphyrins. These data derived from calculation meet well with those from UV–vis spectra and electrochemistry. It is worthy to note that each molecular orbital in these multi(phenylthio)porphyrins marginally derives from either a1u or a2u symmetry. Although identifying the symmetry of FMO from a1u or a2u symmetry is not always easy, it is still possible to judge that HOMOs in phenylthioporphyrins do not always correspond to a2u-like orbitals as that in 11, an order inversion in energy of a2u-like and a1u-like orbitals is often observed in multi-(phenylthio)porphyrins (Fig. 3, 2-S and 5-S as exceptions).

|
Download:
|
| Fig. 3. Orbital energy levels of 11 and phenylthioporphyrins. | |
{kind=link}
In summary we developed a facile method for the synthesis of multi-(phenylthio)porphyrins from multi-halogenated porphyrins and PhSNa. All of these products are well characterized by 1H NMR, 13C NMR and HRMS. Structures of three typical molecules are unambiguously elucidated by X-ray single crystal diffraction. Their UV–vis spectra reveal that the fixation of phenylthio groups usually leads to bathochromic shift. The shift values are highly depended on the amount, position and mutual interactions of phenylthio groups. Electrochemical data meet well with the UV–vis data and indicate that phenylthio groups can be applied to modulate the redox potentials of porphyrins due to their electron-donating effect. DFT calculation further confirms the conclusion above and indicates that an inversion of the order of a2u-like and a1u-like orbitals in energy is common in multi-(phenylthio)porphyrins.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Nature Science Foundation of China (Nos. 21702057, 21772036, and 21602058), the Scientific Research Fund of Hunan Provincial Science and Technology Department (No. 2017JJ3199), Science and Technology Planning Project of Hunan Province (No. 2018TP1017) and the Opening Fund of Key Laboratory of Chemical Biology and Traditional Chinese Medicine Research (Ministry of Education of China).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.022.
| [1] |
(a) K.M. Kadish, K.M. Smith, R. Guilard, The Porphyrin Handbook San Diego, (2000); (b) C. Li, J. Zhang, J. Song, Y. Xie, J. Jiang, Sci. China Chem. 61 (2018) 511-514; (c) W. Miao, Z. Zhu, Z. Li, E. Hao, L. Jiao, Chin. Chem. Lett. 30 (2019) 1895-1902. |
| [2] |
(a) K. Zeng, Z. Tong, L. Ma, et al., Energy Environ. Sci. 13 (2020) 1617-1657; (b) A. Mahmood, J.Y. Hu, B. Xiao, et al., J. Mater. Chem. A 6 (2018) 16769-16797; (c) M. Urbani, M. Grätzel, M.K. Nazeeruddin, T. Torres, Chem. Rev. 114 (2014) 12330-12396; (d) L.L. Li, E.W.G. Diau, Chem. Soc. Rev. 42 (2013) 291-304; (e) Y. Rio, P. Vázquez, E. Palomares, J. Porphyrins Phthalocyanines 13 (2009) 646-651. |
| [3] |
(a) M. Pawlicki, H.A. Collins, R.G. Denning, H.L. Anderson, Angew. Chem. Int. Ed. 48 (2009) 3244-3266; (b) N. Aratani, D. Kim, A. Osuka, Chem. Asian J. 4 (2009) 1172-1182; (c) K.S. Kim, J.M. Lim, A. Osuka, D. Kim, J. Photochem. Photobiol. C 9 (2008) 13-28. |
| [4] |
(a) J. Tian, B. Huang, M.H. Nawaz, W. Zhang, Coord. Chem. Rev. 420 (2020) 213410-213429; (b) S.M.M. Lopes, M. Pineiro, T.M.V.D. Pinho e Melo, Molecules 25 (2020) 3450-3488; (c) R.D. Teo, J.Y. Hwang, J. Termini, Z. Gross, H.B. Gray, Chem. Rev. 117 (2017) 2711-2729; (d) M. Ethirajan, Y. Chen, P. Joshi, R.K. Pandey, Chem. Soc. Rev. 40 (2011) 340-362. |
| [5] |
M. Berthelot, G. Hoffmann, A. Bousfiha, et al., Chem. Commun. (Camb.) 54 (2008) 5414-5417. DOI:10.1039/C8CC01375F |
| [6] |
R.A. Binstead, M.J. Crossley, N.S. Hush, Inorg. Chem. 30 (1991) 1259-1264. DOI:10.1021/ic00006a019 |
| [7] |
M.S. Liao, S. Scheiner, Chem. Phys. Lett. 367 (2003) 199-206. DOI:10.1016/S0009-2614(02)01700-1 |
| [8] |
(a) L.L. Yang, X.L. Hu, Z.Q. Tang, X.F. Li, Chem. Lett. 44 (2015) 1515-1517; (b) D.M. Shen, C. Liu, X.G. Chen, Q.Y. Chen, J. Org. Chem. 74 (2009) 206-211. |
| [9] |
(a) A. Jana, M. Ishida, K. Kwak, et al., Chem. Eur. J. 19 (2013) 338-349; (b) K.A. Nielsen, E. Levillain, V.M. Lynch, J.L. Sessler, J.O. Jeppesen, Chem. Eur. J. 15 (2009) 506-516; (c) H. Li, J.O. Jeppesen, E. Levillain, J. Becher Chem. Commun. (2003) 846-847; (d) J. Becher, T. Brimert, J.O. Jeppesen, et al., Angew. Chem. Int. Ed. 40 (2001) 2497-2500; (e) K.I. Sugiura, K. Ushiroda, M.T. Johnson, J.S. Miller, Y. Sakata, J. Mater. Chem. 10 (2000) 2507-2514; (f) K.I. Sugiura, M.R. Kumar, T.K. Chandrashekar, Y. Sakata, Chem. Lett. 26 (1997) 291-292. |
| [10] |
(a) N. Fukui, A. Osuka, Bull. Chem. Soc. Jpn. 91 (2018) 1131-1137; (b) K. Fujimoto, H. Yorimitsu, A. Osuka, Org. Lett. 16 (2014) 972-975; (c) A.A. Ryan, S. Plunkett, A. Casey, T. McCabe, M.O. Senge, Chem. Commun. (Camb. ) 50 (2014) 353-355; (d) G.Y. Gao, A.J. Colvin, Y. Chen, X.P. Zhang, J. Org. Chem. 69 (2004) 8886-8892. |
| [11] |
(a) H.C. Sample, M.O. Senge, Eur. J. Org. Chem. 2021 (2021) 7-42; (b) M. Kielmann, K.J. Flanagan, K. Norvaiša, D. Intrieri, M.O. Senge, J. Org. Chem. 82 (2017) 5122-5134; (c) Q. Chen, Y.Z. Zhu, Q.J. Fan, S.C. Zhang, J.Y. Zheng, Org. Lett. 16 (2014) 1590-1593; (d) M.J. Crossley, P.L. Burn, S.S. Chew, F.B. Cuttance, I.A. Newsom, J. Chem. Soc. Chem. Commun. (1991) 1564-1566; (e) F. Ma, L. Zhou, Q. Liu, C. Li, Y. Xie, Org. Lett. 21 (2019) 733-736. |
| [12] |
N. Fukui, H. Yorimitsu, A. Osuka, Angew. Chem. Int. Ed. 54 (2015) 6311-6314. DOI:10.1002/anie.201501149 |
| [13] |
(a) A. Tsuda, H. Furuta, A. Osuka, J. Am. Chem. Soc. 123 (2001) 10304-10321; (b) D. Shimizu, K. Fujimoto, A. Osuka, Angew. Chem. Int. Ed. 57 (2018) 9434-9438; (c) K. Kato, K. Furukawa, A. Osuka, Chem. Eur. J. 24 (2018) 572-575. |
| [14] |
(a) K. Fujimoto, H. Yorimitsu, A. Osuka, Org. Lett. 16 (2014) 972-975; (b) K. Fujimoto, H. Yorimitsu, A. Osuka, Org. Lett. 16 (2014) 3172. |