 2021, Vol. 32
2021, Vol. 32 


b College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China
Ynamide is a type of N-substituted electron rich alkynes which exhibits unique chemical properties and serves as flexible synthons in organic synthesis [1]. Therefore, the development of divergent transformations for ynamides has gained considerable attention [2]. In typical, their oxofunctionalization was widely investigated since this process could deliver privileged functionalized amides. For example, Liu and Ye reported that the oxofunctionalization of ynamides could be achieved upon transition-metal-catalysis with pyridine- N-oxide [3]. Meanwhile, the addition reaction of ynamides with carboxylic acids could trigger oxofunctionalization [4]. This has been successfully applied in racemization-free ligation of amides, peptides and macrocyclic esters by Zhao and coworkers [5]. Besides, an oxoallylation of ynamide with allyl alcohol has been nicely established by Ye, and this process could produce privileged medium-sized lactams in an efficient manner [6]. Liu reported a PtCl2-catalyzed oxoarylation of terminal ynamides with nitrones [7], while the internal ynamides could not achieve oxoarylation in this process [8]. Therefore, the investigation of oxoarylation for both terminal and internal ynamides toward privileged molecules construction remains challenging and important.
N-Aryl hydroxamic acids exist extensively in naturally occurring secondary metabolites and these compounds exhibit weak acidities [9]. They are known to be used as metal-chelating agents in biological investigation and directing group in metal-catalyzed tansformation [10-12]. In typical, Maulide reported a sigmatropic rearrangement of hydroxamic acid, and this process involved an addition reaction between hydroxamic acid and the in situ generated electrophilic enolonium-like intermediate [13]. Moreover, they also tested the addition reaction of hydroxamic acid with oxazolidinone-derived ynamide to give an oxoarylation product in 30% yield upon treatment with triflimide. This indicated that the oxoarylation of ynamides with hydroxamic acids would be promising. In continuation of our interests in ynamides [14], we hypothesized that the utilization of transition-metal-catalyst might promote their oxoarylation process in an efficient manner. Herein, we would like to report a Cu(OTf)2-catalyzed oxoarylation of ynamides with hydroxamic acids for facile and selective entry to (ortho-aminoaryl)amides and oxindoles.
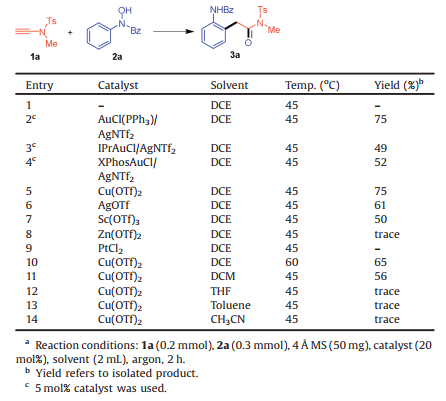
We commenced our study by investigating N-sulfonyl ynamide 1a and N-benzoyl- N-phenylhydroxylamine 2a. Initially, we conducted the reaction in DCE at 45 ℃ without any catalyst and there was no new product detected (Table 1, entry 1). When AuCl(PPh3)/AgNTf2 was used as catalyst (5 mol%), and gratifyingly, a new product 3a was observed and isolated in 75% yield (entry 2). The standard analysis including 1H NMR, 13C NMR and HRMS showed that 3a was an (ortho-aminoaryl)amide compound. This indicated an oxoarylation occurrence. Variations of the catalysts to IPrAuCl/AgNTf2 and XPhosAuCl/AgNTf2 would decrease the yield to 49% and 52% respectively (entries 3 and 4). The next survey of catalysts showed that Cu(OTf)2 could deliver 3a in a comparable 75% yield (entry 5), while the use of AgOTf and Sc(OTf)3 would lead to lower yields (entries 6 and 7). To our surprise, the utilization of Zn(OTf)2 would only give trace product (entry 8). When PtCl2 was used, the hydrolyzation rather than oxoarylation was observed (entry 9). On the other hand, either increasing the temperature to 60 ℃ with Cu(OTf)2 catalyst or changing the solvent to DCM at 45 ℃ would decrease the yield (entries 10 and 11). Moreover, the survey of solvents showed that THF, toluene and CH3CN would shut down the reactivity to deliver trace product (entries 12–14).
|
|
Table 1 Reaction conditions.a |
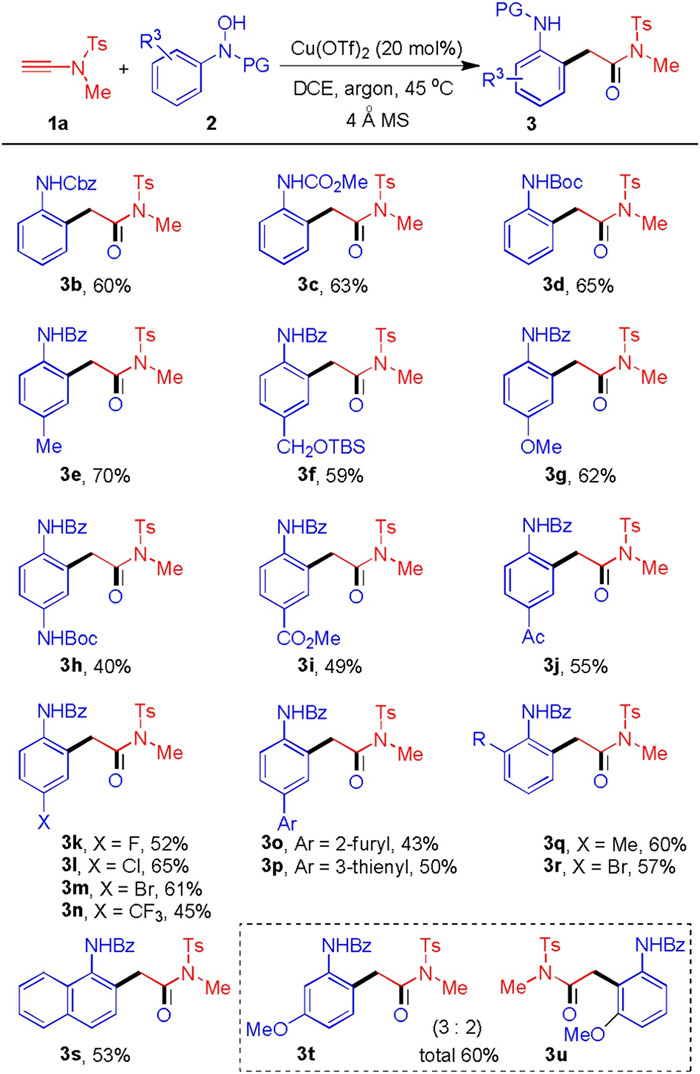
With the optimized reaction condition in hand, we next tested the substrate scope (Scheme 1). As shown in Scheme 1, the N-protecting groups of hydroxamic acids varied from benzoyl to Cbz, ester and Boc, and these hydroxamic acids could serve as oxoarylation source for ynamide 1a to give the products in good yields (3b-3d). Meanwhile, many functional groups substituted on the aromatic ring of N-aryl hydroxamic acids were tested. For example, the para-substituted N-aryl hydroxamic acids could well engage in this cascade process to deliver the corresponding (ortho-aminoaryl)amides in good yields (3e-3j). The functionalities including methyl, hydroxymethyl, methoxy, N-Boc amine, ester, ketone, were well tolerated in this process and also offered ample opportunities for further derivatization. Moreover, the structure of 3e was confirmed by X-ray analysis (CCDC: 2021769). On the other hand, the halogen substitutions like fluoro, chloro, bromo, and CF3, were compatible in this reaction (3k-3n). The heterocyclic substitutions like furan and thiophene could also perform well in this process (3o and 3p). Meanwhile, the ortho-substituted N-aryl hydroxamic acids were employed in this oxoarylation to furnish the products smoothly (3q and 3r). When the N-(naphthalen-1-yl)hydroxamic acid 1s was used, the regioselective (naphthalen-1-amino-2-yl)amide 3s could be formed exclusively. When the meta-substituted N-aryl hydroxamic acid 2t was subjected, the process would deliver a mixture of oxoarylation products 3t and 3u (total 60% yield). The ratio of 3t and 3u was 3:2, probably because the process would favor the less steric transition state and the corresponding product 3t was the major isomer.

|
Download:
|
| Scheme 1. Substrate scope of hydroxamic acid. Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), 4 Å MS (50 mg), Cu(OTf)2 (20 mol%), DCE (2 mL), 45 ℃, argon, 2 h. Yields refer to isolated products. | |
{kind=link}
Meanwhile, the generality of ynamides were also explored. As shown in Scheme 2, the terminal ynamides with N-substitutions like isobutyl, furan-2-methyl, (S)-1-phenylethyl, could well engage in this process (4a-4c). And the functionalities like hydroxy, prenyl, cyclohexenyl, were tolerated (4d-4f) and also offer considerable potential for late-stage transformation. On the other hand, the sulfonyl substitutions were investigated while thiophene, phenyl and 4-nitrophenyl were found compatible (4g-4i). Furthermore, the internal ynamides were next subject to this oxoarylation process. As shown in Scheme 2, the substitutions like butyl, cyclopropyl, phenylethyl, allyl, hydroxy, ester, were well applicable to yield the functionalized products smoothly (4j-4o). The aryl substituted ynamides diverged from the reactivity pattern and were observed with hydrolyzation products. Therefore, this process represents a general oxoarylation for both terminal and internal ynamides, and provides a distinct approach to (ortho-aminoaryl)amides.

|
Download:
|
| Scheme 2. Substrate scope of ynamides. Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol), 4 Å MS (50 mg), Cu(OTf)2 (20 mol%), DCE (2 mL), 45 ℃, argon, 2 h. Yields refer to isolated products. | |
{kind=link}
Considering that the moiety of N-tosyl of amides 3 and 4 could act as leaving group in intramolecular cyclization, we envisioned that treatment of 3 and 4 with base might enable a lactamization. Upon completion of the Cu(OTf)2-catalyzed oxoarylation of ynamide 1a and hydroxamic acid 2a, we treated the reaction solution with K2CO3 at 45 ℃. To our delight, we indeed found the consumption of (ortho-aminoaryl)amides and a new product 5a was isolated in 72% yield. The standard analysis showed that 5a was an oxindole compound. We next investigated the scope of this oxoarylation/lactamization process in a one-pot manner. As shown in Scheme 3, a variety of ynamides and hydroxamic acids were subject to this reaction. Besides benzoyl groups, differential protecting groups of hydroxamic acids including Cbz, ester and Boc, were all amenable to this process to deliver the N-protected oxindoles in good yields (5b-5d). The structure of 5c was confirmed by X-ray analysis (CCDC: 2021773). The para-substitutions of hydroxamic acids with methyl, hydroxymethyl, methoxy, amino, ester, ketone, fluoro, chloro, bromo, trifluoromethyl, furan, thiophene, were well tolerated in this one-pot process (5e-5p). Moreover, this process could also produce the 7-substituted and naphthalene-containing oxindoles in moderate yields (5q-5s), which were difficult to be synthesized in conventional methods. Meanwhile, when the internal ynamides were used, this process could deliver the corresponding oxindole products (5t-5y) rapidly. Considering the wealth of oxindoles in medicinal chemistry [15, 16], this method provides a direct approach to these molecules in an efficient way.

|
Download:
|
| Scheme 3. Scope of oxoarylation/lactamization. Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), 4 Å MS (50 mg), Cu(OTf)2 (20 mol%), DCE (2 mL), 45 ℃, argon, 2 h, then K2CO3 (0.4 mmol), 45 ℃, 2 h. Yields refer to isolated products. | |
{kind=link}
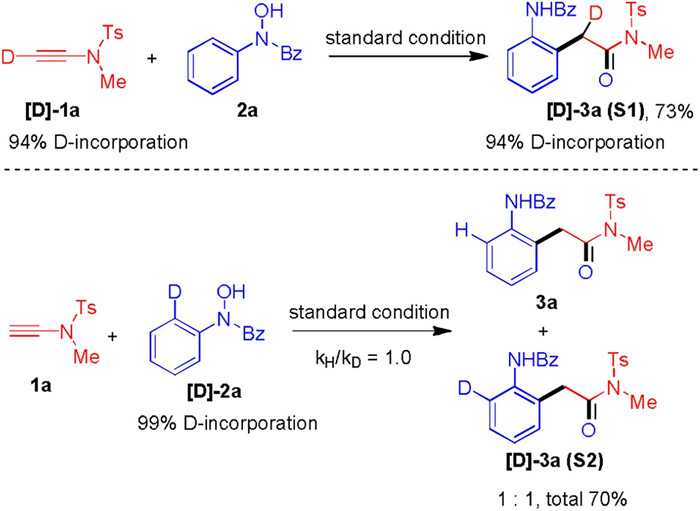
In order to probe the mechanism, a deuterium-labelling reaction was conducted (Scheme 4). The [D]-1a was prepared and subject to reaction with 2a, and [D]-3a was obtained in 73% yield and deuterium is incorporated in the α-position of amide. Additionally, the [D]-2a was prepared and subject to an intramolecular competition experiment to probe the C–H functionalization event. The result showed that a primary kinetic isotope effect was not observed. This suggests that C–H functionalization is not the rate-determining step of this oxoarylation process.

|
Download:
|
| Scheme 4. Isotope labelling experiments. | |
{kind=link}
Based on these results, a plausible reaction mechanism was proposed (Scheme 5). Initially, the ynamides was activated by Cu(OTf)2, which could be attacked by N-aryl hydroxamic acid to deliver intermdiate B. The following rapid [3,3] sigmatropic rearrangement would deliver (ortho-aminoaryl)amide (3 and 4) to constitute the oxoarylation of ynamides. Upon treatment with K2CO3, the (ortho-aminoaryl)amide (3 or 4) would undergo a lactamization to deliver the cyclized oxindoles 5. Therefore, the deuterium atom of [D]-1a would be incorporated on the α-methylene of amide, and the deuterium of [D]-2a did not show difference with H-atom in the deprotonation of [3,3] sigmatropic rearrangement.

|
Download:
|
| Scheme 5. Proposed reaction mechanism. | |
{kind=link}
In summary, an oxoarylation of ynamides with hydroxamic acids has been successfully developed. Both the terminal and internal ynamides could undergo addition/[3,3] sigmatropic rearrangement with hydroxamic acids to constitute an oxoarylation process. This method would produce (ortho-aminoaryl)amides and oxindoles with mild reaction condition, broad substrate scope and high efficiency.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentWe are grateful for the financial support by the National Natural Science Foundation of China (Nos. 21878264, 21971222).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.054.
| [1] |
(a) C.A. Zificsak, J.A. Mulder, R.P. Hsung, C. Rameshkumar, L.L. Wei, Tetrahedron 57 (2001) 7575-7606; (b) X.N. Wang, H.S. Yeom, L.C. Fang, et al., Acc. Chem. Res. 47 (2014) 560-578; (c) F. Pan, C. Shu, L.W. Ye, Org. Biomol. Chem. 14 (2016) 9456-9465; (d) G. Evano, N. Blanchard, G. Compain, et al., Chem. Lett. 45 (2016) 574-585. |
| [2] |
(a) R. Pirwerdjan, P. Becker, C. Bolm, Org. Lett. 18 (2016) 3307-3309; (b) F.L. Hong, Z.S. Wang, D.D. Wei, et al., J. Am. Chem. Soc. 141 (2019) 16961-16970; (c) Y. Xu, Q. Sun, T.D. Tan, et al., Angew. Chem. Int. Ed. 58 (2019) 16252-16259; (d) Z. Zeng, H. Jin, M. Rudolph, et al., Angew. Chem. Int. Ed. 57 (2018) 16549-16553; (e) Q. Zhao, D.F.L. Rayo, D. Campeau, et al., Angew. Chem. Int. Ed. 57 (2018) 13603-13607; (f) Y. Wang, L.J. Song, X. Zhang, J. Sun, Angew. Chem. Int. Ed. 55 (2016) 9704-9708; (g) S.N. Karad, R.S. Liu, Angew. Chem. Int. Ed. 53 (2014) 9072-9076; (h) C. Theunissen, B. Métayer, N. Henry, et al., J. Am. Chem. Soc. 136 (2014) 12528-12531; (i) M. Lecomte, G. Evano, Angew. Chem. Int. Ed. 55 (2016) 4547-4551; (j) D.V. Patil, S.W. Kim, Q.H. Nguyen, et al., Angew. Chem. Int. Ed. 56 (2017) 3670-3674; (k) P. Thilmany, G. Evano, Angew. Chem. Int. Ed. 59 (2020) 242-246; (l) X. Zeng, J. Li, C.K. Ng, et al., Angew. Chem. Int. Ed. 57 (2018) 2924-2928. |
| [3] |
(a) D. Vasu, H.H. Hung, S. Bhunia, et al., Angew. Chem. Int. Ed. 50 (2011) 6911-6914; (b) L. Li, B. Zhou, Y.H. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 8245-8249. |
| [4] |
(a) S. Xu, J. Liu, D. Hu, X. Bi, Green Chem. 17 (2015) 184-187; (b) D.L. Smith, W.R.F. Goundry, H.W. Lam, Chem. Commun. 48 (2012) 1505-1507; (c) H. Liu, Y. Yang, J. Wu, et al., Chem. Commun. 52 (2016) 6801-6804. |
| [5] |
(a) L. Hu, S. Xu, Z. Zhao, et al., J. Am. Chem. Soc. 138 (2016) 13135-13138; (b) M. Yang, X. Wang, J. Zhao, ACS Catal. 10 (2020) 5230-5235. |
| [6] |
(a) B. Zhou, L. Li, X.Q. Zhu, et al., Angew. Chem. Int. Ed. 56 (2017) 4015-4019; (b) B. Zhou, Y.Q. Zhang, K. Zhang, et al., Nat. Commun. 10 (2019) 3234-3244. |
| [7] |
B.S. Bhunia, C.J. Chang, R.S. Liu, Org. Lett. 14 (2012) 5522-5525. |
| [8] |
A. Mukherjee, R.B. Dateer, R. Chaudhuri, et al., J. Am. Chem. Soc. 133 (2011) 15372-15375. DOI:10.1021/ja208150d |
| [9] |
(a) M.J. Miller, Chem. Rev. 89 (1989) 1563-1579; (b) J.B. Neilands, J. Biol. Chem. 270 (1995) 26723-26726; (c) R.C. Hider, X. Kong, Nat. Prod. Rep. 27 (2010) 637-657. |
| [10] |
(a) E. Nuti, D. Cuffaro, E. Bernardini, et al., J. Med. Chem. 61 (2018) 4421-4435; (b) C. Rouanet-Mehouas, B. Czarny, F. Beau, et al., J. Med. Chem. 60 (2017) 403-414. |
| [11] |
(a) H.Y. Wang, L.L. Anderson, Org. Lett. 15 (2013) 3362-3365; (b) J. Wen, A. Wu, P. Chen, et al., Tetrahedron Lett. 56 (2015) 5282-5286. |
| [12] |
(a) C. Beshara, A. Hall, R. Jenkins, et al., Org. Lett. 7 (2005) 5729-5732; (b) A. Porzelle, M. Woodrow, N. Tomkinson, Eur. J. Org. Chem. 2008 (2008) 5135-5143; (c) A. Porzelle, M. Woodrow, N. Tomkinson, Org. Lett. 12 (2010) 812-815; (d) A. Porzelle, M. Woodrow, N. Tomkinson, Org. Lett. 12 (2010) 1492-1495. |
| [13] |
S. Shaaban, V. Tona, B. Peng, N. Maulide, Angew. Chem. Int. Ed. 56 (2017) 10938-10941. DOI:10.1002/ange.201703667 |
| [14] |
(a) B. Huang, L. Zeng, Y. Shen, S. Cui, Angew. Chem. Int. Ed. 56 (2017) 4565-4568; (b) Y. Shen, B. Huang, L. Zeng, S. Cui, Org. Lett. 19 (2017) 4616-4619; (c) R. Chen, Y. Liu, S. Cui, Chem. Commun. 54 (2018) 11753-11756; (d) L. Zeng, H. Sajiki, S. Cui, Org. Lett. 21 (2019) 6423-6426; (e) C. Chen, S. Cui, J. Org. Chem. 84 (2019) 12157-12164; (f) L. Zeng, Z. Lai, C. Zhang, et al., Org. Lett. 22 (2020) 2220-2224; (g) R. Chen, L. Zeng, B. Huang, et al., Org. Lett. 20 (2018) 3377-3380; (h) Y. Shen, C. Wang, W. Chen, et al., Org. Chem. Front. 5 (2018) 3574-3578. |
| [15] |
(a) A.B. Dounay, L.E. Overman, Chem. Rev. 103 (2003) 2945-2964; (b) J.J. Badillo, N.V. Hanhan, A.K. Franz, Curr. Opin. Drug Disc. 13 (2010) 758-776; (c) R. Dalpozzo, G. Bartoli, G. Bencivenni, Chem. Soc. Rev. 41 (2012) 7247-7290; (d) P. Fu, F. Kong, X. Li, et al., Org. Lett. 16 (2014) 3708-3711; (e) J. Zhang, Z. Qian, X. Wu, et al., Org. Lett. 16 (2014) 2752-2755. |
| [16] |
(a) M. Chen, X. Wang, P. Yang, et al., Angew. Chem. Int. Ed. 59 (2020) 12199-12205; (b) W. Kong, Q. Wang, J. Zhu, J. Am. Chem. Soc. 137 (2015) 16028-16031; (c) L. Yin, M. Kanai, M. Shibasaki, Angew. Chem. Int. Ed. 50 (2011) 7620-7623; (d) J. Wang, Y. Yuan, R. Xiong, et al., Org. Lett. 14 (2012) 2210-2213; (e) M. Ratushnyy, N. Kvasovs, S. Sarkar, et al., Angew. Chem. Int. Ed. 59 (2020) 10316-10320; (f) P. Fan, Y. Lan, C. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 2180-2186; (g) X. Bai, C. Wu, S. Ge, Y. Lu, Angew. Chem. Int. Ed. 59 (2020) 2764-2768; (h) J. Zhu, L. Huang, W. Dong, et al., Angew. Chem. Int. Ed. 58 (2019) 16119-16123; (i) Z.J. Zhang, L. Zhang, R.L. Geng, et al., Angew. Chem. Int. Ed. 58 (2019) 12190-12194. |