 2021, Vol. 32
2021, Vol. 32 


b School of Civil and Enviro nmental Engineering, Ningbo University, Ningbo 315211, China;
c Technology Innovation Center for Land Spatial Eco-Restoration in Metropolitan Area, Ministry of Natural Resources, Shanghai 200062, China;
d Shanghai Engineering Research Center of Biotransformation of Organic Solid Waste, Shanghai 200241, China;
e Jiangsu Provincial Key Laboratory of Enviro nmental Science and Engineering, Suzhou University of Science and Technology, Suzhou 215009, China
In recent years, advanced oxidation processes (AOPs), as an attractive and effective new oxidation technology, has been extensively investigated in the field of water purification. AOPs have the advantages of wide application range, fast reaction rate and strong oxidizability. Chloride ion (Cl−) is ubiquitous in the industrial wastewater, and it is an important influencing factor affecting the overall treatment performance of AOPs [1, 2]. Recently, the involvement of Cl− in the saline wastewater treatment with sulfate radical (SO4·‒)-based AOPs has received widespread attention. However, the effects of Cl− in different AOPs are still controversial so far, due to the complicated reaction processes. For instance, Anipsitakis et al. [3] found the accelerated degradation kinetics of phenol and 2, 4-dichlorophenol with the presence of a small amount of Cl− in the cobalt/peroxymonosulfate (Co/PMS) system. Chan and Chu [4] reported that the degradation efficiency and reaction rate of Co-mediated PMS oxidation process were significantly inhibited in the presence of Cl−. Conversely, in our previous studies, a dual effect of Cl− (inhibitory then promoting) was observed in oxidative degradation of aromatic compounds by the PMS-based AOPs systems [5-9]. At low concentrations, the inhibition of Cl− was attributed to the scavenging of SO4·‒ and consequent production of less reactive chlorine radicals (Cl2·‒, Cl·, and ClOH·‒) (Eqs.1-7). While high concentrations of chloride were found to react directly with PMS through a non-radical pathway to generate main oxidative species HClO/Cl2 (Eqs. 8 and 9), leading to a rapid degradation of organic contaminants. In addition, Yuan et al. [10, 11] reported another dual effect of Cl‒ (promoting then inhibitory) in degradation of acid orange 7 (AO7) in the ultraviolet/peroxydisulfate (UV/PDS) and UV/TiO2 systems through chain-transfer mechanisms involving chlorine radicals and changes of surface adsorption of dyes on TiO2. This dual effect has also been reported in the oxidative degradation of diclofenac by heat-activated PDS [12]. No significant effects of Cl− (0−300 mmol/L) on the degradation of monochlorophenols were observed during UV/PDS oxidation [13], but the presence of Cl− greatly inhibited the degradation of polychlorinated biphenyls (PCBs) in the UV/PDS system [14]. However, Criquet and Leitner [15] found that the addition of Cl− facilitated the degradation of acetic acid by UV/PDS. In conclusion, for different target compounds, the impacts of Cl− in the same UV/PDS system vary.
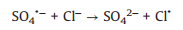
|
(1) |

|
(2) |
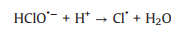
|
(3) |
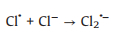
|
(4) |

|
(5) |
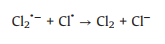
|
(6) |
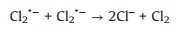
|
(7) |
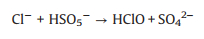
|
(8) |

|
(9) |
In addition to the influence of exogenic Cl− (inorganic form from water matrix) ions in saline wastewater on the treatment processes of AOPs, the importance of endogenic Cl− (organic form from pollutant itself) of chlorinated organic compounds on the oxidation performance has also been recently highlighted [6, 9, 13, 16-20]. Xu et al. [20] revealed that the chlorine atom/ion released by dechlorination during 2, 4, 6-trichlorophenol degradation might involve in electrophilic addition reactions to form polychlorinated compounds (chlorine atom number ≥ 3, such as 2, 4, 5-trichlorophenol and 2, 3, 4, 6-tetrachlorophenol). Our most recent investigation revealed that the position of chlorine in chlorinated compounds has potential impact on their degradation rates. For instance, 3-chlorophenol was more susceptible to oxidative degradation by the UV/PDS system than 4-chlorophenol and 2-chlorophenol, while a higher absorbable organic halogen (AOX) value was observed for 4-chlorophenol [13]. The formation of AOX during halogenated compounds oxidation depends largely on the exogenic Cl−/Br− concentration [19]. At low level of exogenic Cl−/Br−, the generated active halogen species are more prone to electrophilic addition reaction with the benzene ring and result in AOX accumulation. Conversely, at higher exogenic Cl−/Br− concentration, a large amount of active halogen species may mediate the re-dehalogenation reaction of halogenated intermediates and/or carbon skeleton cleavage to reduce halogen incorporation and thus AOX concentration. Note that although several promoting effects of exogenic Cl− have been found in some AOPs, while these treatment processes also accompany with low mineralization rate, undesirable increase of AOX and the formation of chlorinated byproducts with high toxicity [8, 13, 17, 19, 20].
With the rapid development of dye production and related industries, dyeing wastewater has become one of the recognized major sources of industrial pollution. In the dye family, there are many dyes with chlorine atom(s) in the molecular structure and some chlorinated organic dyes are listed in Table S1 (Supporting information). Among them, acid yellow 17 (AY-17), a typical monoazo dye with two chlorine in the molecular structure, is widely used in textile, food, paper and cosmetic industries. It is also commonly used as an additive in ordinary household products such as personal care, soap, dishwashing liquid, and alcoholic perfumes [21]. The AY-17 dye present in the water enviro nment can be partially removed through adsorption, hydrolysis, and photolysis, and produce more toxic aromatic amines, which can harm the respiratory and nervous systems of humans and organisms [21, 22]. But most AY-17 dyes still exist in the water enviro nment in the form of precursor compounds. Therefore, it is of great practical significance to study the effective removal method of AY-17 dyes and its transformation pathway. Some researchers have conducted studies on the degradation of AY-17 by AOPs under a wide range of experimental conditions, and confirmed the effective performance of the AOPs [23-27]. However, previous studies on AY-17 mainly focused on the degradation process under the hydroxyl radical (·OH)-based AOPs, and there are few reports about the degradation of AY-17 by SO4·−-involved AOPs. There is no comprehensive study on the AOX evaluation and chlorinated byproducts analysis, and there are still some deficiencies and controversies in the degradation mechanisms. Khan et al. [24] proposed that the photo-Fenton system achieved the degradation of AY-17 through dechlorination, desulfurization, addition reaction and dehydrogenation to produce ketone-containing or/and oxygen-containing six-membered ring intermediates, without generation of chlorinated compounds. However, the photoelectrocatalytic degradation products in the report of Huda et al. [23] were mainly chlorinated aromatic compounds and benzenesulfonic acid compounds formed via −N=N− cleavage and hydroxyl addition reaction.
In this work, AY-17 was selected as a model chlorinated pollutant to systematically evaluate the impact of exogenic Cl− on the degradation kinetics. The effects of initial pH, oxidant content, dye concentration, common ions on AY-17 photooxidative degradation in the UV/PDS system were investigated. Liquid chromatography-mass spectrometry (LC–MS/MS) was used to identify the degradation products in the absence and presence of exogenic Cl−.
Chemicals and reagents and experimental procedure are shown in Texts S1 and S2 (Supporting information). The UV–vis spectrophotometer operating from 200 nm to 900 nm was used for the quantitative analysis of AY-17 dye solution by measuring the absorbance at various time intervals. The maximum absorption peak of AY-17 is located at λmax = 402 nm, corresponding to the −N=N− group of azo dye. The total organic carbon (TOC) concentration of samples were measured by TOC analyzer (multi N/C 3100, Analytic Jena, Germany). Absorbable organic halogen were determined by microcoulometry using an AOX analyzer (multi X 2500, Analytic Jena, Germany). The detection range of AOX analysis was 1−100 μg/L (calculated as the absolute content of organic chloride). The ultra high liquid chromatography and mass spectrometry (UPLC-Q-tof-MS) equipped with an Agilent 1290 ultra-high-performance liquid chromatography system and a high-resolution quadrupole time-of-flight mass spectrometer (Agilent 6540 QTOF) was used to identify the intermediate products generated during AY-17 degradation process by UV/PDS. The product test was performed using a flying mass spectrometer in positive ionization mode, namely ESI (+) was an ion source with a voltage of 3.5 kV. And more information about UPLC-Q-tof-MS analysis are available in our previous study [28].
The preliminary experiments indicated that the direct oxidation of AY-17 by PDS itself in the dark or direct photolysis of the dye without PDS was negligible. However, AY-17 could be decolorized effectively in the presence of both UV light and PDS. The photodegradation reaction of AY-17 follows pseudo-first order kinetics and the fitting coefficients R2 are shown in Table S2 (Supporting information).
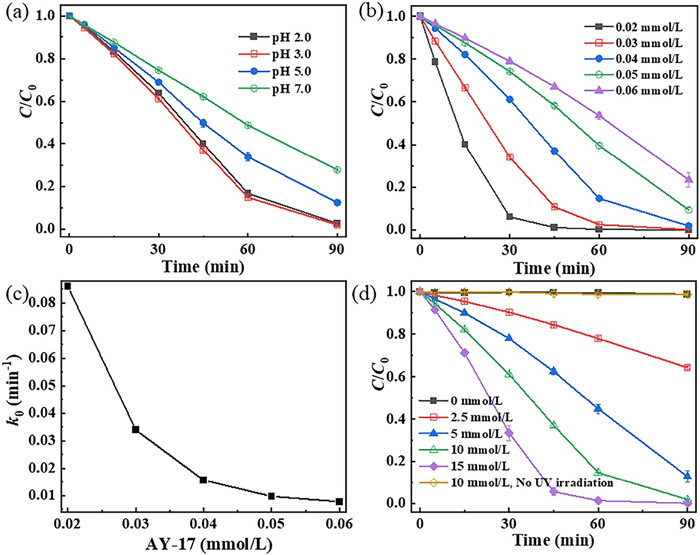
It is known that pH does not only affect the formation and redox potential of active radicals in AOPs processes [17, 29], but may also affect the dissociation of pollutants [27]. The alkaline enviro nment is known to be favorable for persulfate (PS) activation [30, 31]. Accordingly, to exclude the effect of alkali activation, the effect of initial solution pH on AY-17 decolorization was determined at pH 2.0, 3.0, 5.0 and 7.0, respectively. As shown in Fig. 1a, it indicates that AY-17 degradation was significantly influenced by the initial pH, and higher degradation efficiency was achieved at acidic pH value. There was no significant difference in the degradation efficiency of AY-17 at pH 2.0 and 3.0. However, when the pH was further increased to 5.0 and 7.0, the dye degradation was apparently inhibited. According to previous reports, the degradation efficiency of AY-17 in the UV/TiO2 [32] and UV/H2O2/Fe2+ [24] systems reached the maximum at pH 3.0, which was 70.6% after 375 min and 88% in 25 min, respectively. In this work, pH 3.0 with a degradation efficiency of 98.2% was selected as a control pH for the subsequent photochemical oxidation experiments.

|
Download:
|
| Fig. 1. The decolorization rate of AY-17 as a function of (a) pH, (b) substrate concentration and (c) the relationship between kinetic rate constants and AY-17 concentrations, and (d) oxidant content. | |
It is generally recognized that sulfate radicals could react with water molecules to generate hydroxyl radicals (Eq. 10) [33, 34]. In the UV/PDS system, both SO4·− and ·OH radicals were reactive species responsible for contaminants degradation. It is reported that SO4·‒ possess higher redox potential (2.5–3.1V), longer life time than·OH and more selective for pollutants oxidation [35]-37]. SO4·‒ radicals are the primary reactive species for AY-17 degradation at acidic enviro nment, while the increase of pH renders the transformation of SO4·‒ into ·OH (Eq. 11) [38, 39]. This explains the decreased decolorization efficiency at higher pH. Moreover, under acidic conditions, S2O82‒ molecules could be easily catalyzed by acids and further decomposed to produce more SO4·‒ (Eqs. 12 and 13) [40, 41].

|
(10) |
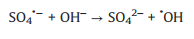
|
(11) |
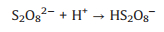
|
(12) |

|
(13) |
The effect of AY-17 concentration (0.02−0.06 mmol/L) on the photooxidative degradation was determined with 10 mmol/L PDS at pH 3.0, as shown in Fig. 1b. The decolorization efficiency decreased with an increasing amount of AY-17 concentration with a constant concentration of PDS, similar to previous studies [42]. 0.02 mmol/L AY-17 could be almost completely degraded (99.8%) in 60 min, while the degradation rate of 0.06 mmol/L AY-17 decreased to 76.6% at 90 min. The degradation rate constants of dye from 0.02 mmol/L to 0.06 mmol/L were 8.61 × 10−2 min-1, 3.39 × 10‒2 min‒1, 1.56 × 10‒2 min‒1, 9.68 × 10‒3 min‒1 and 7.69 × 10‒3 min‒1, respectively (Fig. 1c).
The phenomenon mentioned above may be explained by the following reasons. With the increasing concentration of AY-17, the amount of AY-17 molecules in solution increased, but the level of SO4·− remained constant. Therefore, an increase in dye concentration led to a lower degradation efficiency of AY-17. On the other hand, high concentration of AY-17 was likely to reduce the absorption of UV light by PDS, due to the inner filtering effect [26, 43]. In that case, the reaction of PDS photolysis to form SO4·− radicals may also be restrained. Moreover, reactive radicals might be captured by the degradation byproducts or the excess S2O82‒ molecules at low AY-17 concentration (Eqs. 14 and 15) [44-46], but this effect became negligible when the concentration of AY-17 was adequate [47].

|
(14) |
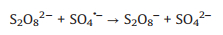
|
(15) |
To examine the effect of oxidant concentration on dye degradation rate, the measurements were carried out at a variable concentrations range of PDS (0−15 mmol/L). As shown in Fig. 1d, the decolorization efficiency of AY-17 in the UV/PDS system was highly dependent on the dosages of PDS. The degradation rate of AY-17 increased significantly as PDS concentration increased, which has been widely recognized [48, 49]. It indicated that the degradation efficiency of AY-17 was 35.7%, 87.1%, 98.2% and 100% corresponding to 2.5 mmol/L, 5 mmol/L, 10 mmol/L and 15 mmol/L PDS after 90 min of the reaction, respectively. Higher PDS concentrations ensured that sufficient reactive radicals were available to enhance photolytic oxidation performance [38, 50], and AY-17 degradation turned into more efficient.
Generally, a large amount of salts are used in various printing and dyeing processes to enhance dyeing, but their presence in dyeing wastewater may affect the oxidative degradation efficiency of dyes. Herein, the effects of several inorganic ions (Cl−, Br−, NO3− and NH4+) that commonly occur in real dyeing wastewater on AY-17 degradation in the UV/PDS system were tested (Fig. 2).

|
Download:
|
| Fig. 2. The effects of different inorganic ions concentrations on AY-17 degradation in the UV/PDS system: (a) Cl−, (b) Br−, (c) NH4+ and (d) NO3−. Experimental conditions: [AY-17]0=0.04 mmol/L, [PDS]0=10 mmol/L and pH 3.0. | |
Figs. 2a-c show that the removal of AY-17 was accelerated by the introduction of different amounts of Cl−, Br‒ and NH4+, but such promoting effect declined gradually when concentrations exceeded certain values. Similar accelerating effects were also found in the UV/PDS the heat/PDS and the UV/TiO2 systems [10-12]. The maximum rate constants of AY-17 were 1.08 × 10-2 min‒1, 9.92 × 10‒3 min‒1 and 1.02 × 10‒2 min‒1 when Cl−, Br− and NH4+ ions concentrations were 15 mmol/L, 0.10 mmol/L and 100 mmol/L, respectively. The removal rate of AY-17 was 98.2% after 90 min in the absence of inorganic ions, whereas the above three inorganic ions could accomplish 98.8%, 97.3%, and 98.7% of dye decolorization in 60 min at the optimum concentrations, respectively. Fig. 2d shows that low concentration (2.94 mmol/L) NO3− exhibited a little inhibitory effect on the decolorization of AY-17. However, when the concentration reached 5.88 mmol/L and above, NO3− could accelerate the dye degradation. The removal rate of AY-17 with 23.5 mmol/L NO3− after 60 min was 96.4%.
SO4·− is mainly responsible for the decolorization of AY-17 when halide ions are not added. The addition of halide ions (Cl‒/Br‒) triggered the conversion of SO4·− radicals and ·OH radicals to more selective halogen radicals (X·, X2·‒ and XOH·‒) [11, 51]. Subsequently, halogen radicals partially replaced SO4·− to participate in the degradation processes of AY-17, and even became the main active species in the UV/PDS system. Criquet and Leitner [15] reported that the Cl· radicals converted by SO4·− radicals are more reactive with acetate ions, resulting in accelerated degradation of acetic acid. The rate constant for the reaction of Cl· with acetic acid (k=3.7 × 109 L mol‒1 s‒1) is three orders greater than SO4·‒ (k=5.0 × 106 L mol‒1 s‒1). Besides, the halogen radicals probably have better reactivity to AY-17 and/or intermediate byproducts than SO4·−, which counteracted or even exceeded the adverse effects of chloride in competition with dyes and intermediates for SO4·− radicals (Eq. 16). Cl‒ can react with SO4·− to form active chlorine radicals, which may participate in the chain-reactions with PDS to increase the total yield of SO4·− [12]. In addition, inorganic ions and the derived reactive species might reduce the recombination frequency of SO4·− or ·OH (Eqs.16-18) [11], restraining the consumption of oxidizing species. When chloride concentration was high enough, the self-quenching of halogen radicals might occur, which explained the lower promotion effect with increasing concentration. The main reactions involving halide ions could be summarized as Eqs. 1-7 [17, 50, 52].
It should be noted that even in the same oxidation system, the effects of Cl− might different. Fang et al. [14] observed that the presence of Cl− obviously inhibited the degradation of 2, 4, 4'-trichlorobiphenyl in the UV/PDS system, while Cl− showed a dual effect (promoting then inhibitory) on biphenyl degradation. Over a wide range of concentrations (0−300 mmol/L), Cl− had no significant effect on the degradation of monochlorophenols (MCPs, 2-CP, 3-CP and 4-CP), and the value of the rate constant at different chloride concentrations basically remained unchanged [13]. Besides, the effects of exogenic Cl− were slightly different due to the different positions of chlorine in the structure of the substrates. The degradation of 2-CP and 3-CP were promoted by Cl−, but its inhibitory effect was observed for 4-CP degradation. Low levels of Cl− (1−100 mmol/L) and Br− (0.01−0.1 mmol/L) significantly accelerated the removal efficiency of acid orange 7 (AO7) by the UV/PDS system, but this promotion effect gradually decreased with higher amount of halides [11]. In addition, the effect of Cl‒ on organics degradation might also different due to the diverse activation methods for SO4·‒ generation [53]. The results of this study were inconsistent with previous studies, which may be closely related to the selectivity of various reactive species, reaction conditions, the properties and structure of target contaminants, and complicated degradation mechanisms [3, 14].
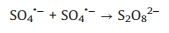
|
(16) |
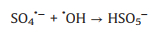
|
(17) |
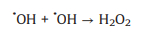
|
(18) |
The effects of some trace metal ions (0.01 mmol/L) on AY-17 degradation were investigated by maintaining constant concentrations of both dye (0.04 mmol/L) and oxidant (2.5 mmol/L). The AY-17 did not degrade with Fe3+ or Ag+ in the presence of PDS alone or by direct photolysis without PDS, even in the dark containing PDS (data not shown). As shown in Fig. 3a, Cu2+ had a little effect on the degradation of AY-17. Cr3+, Mn2+, Co2+ and Ce4+ had negligible promoting effects on dye degradation. While AY-17 decolorization rate was significantly improved with the introduction of Ag+ or Fe3+, and the degradation efficiency in 90 min increased from 35.7% to 62.1% and 81.8%, respectively. Previous studies have confirmed the ability of transition metal ions to participate in PS activation via electron transfer to generate reactive radicals and promote the effective degradation of pollutants [44]. Ag+ possibly acted as a redox catalyst directly participating in AY-17 degradation processes (Eqs. 19-21), which are similar to the oxidation reactions of tartrazine under the PDS/Ag+ system [44]. Ag+ firstly reacted with S2O82− molecule to form Ag2+ and SO4·‒ radical, and the generated reactive radical reacted with Ag+ to produce Ag2+, which was further reduced back to Ag+ by the -N=N- group to ensure the cyclic catalytic reactions. Fe3+ underwent photolysis under ultraviolet irradiation to form Fe2+ and more ·OH radicals, and then the Fe2+ was oxidized by S2O82− molecule to Fe3+ with the generation of SO4·‒ radical (Eqs. 22 and 23) [44, 54]. Besides, the important role of high-valent species (Fe(Ⅳ)) in the Fe(Ⅱ)/PDS system was recently reported (Eq. 24) [55, 56]. This kind of reaction probably also occurred between intermediate Fe2+ and PDS in this system.
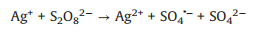
|
(19) |
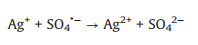
|
(20) |
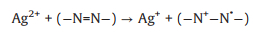
|
(21) |
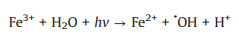
|
(22) |

|
(23) |

|
(24) |

|
Download:
|
| Fig. 3. The effects of trace metal ions (0.01 mmol/L) (a) and oxalic acid concentration (b) on AY-17 photodegradation. Experimental conditions: [AY-17]0=0.04 mmol/L and pH 3.0. | |
In addition, oxalic acid is a common byproduct at the end of the advanced oxidation of many organic contaminants, and it might affect the degradation processes of target pollutants due to its competition for oxidative species and strong complexation capability with transition metals [57-60]. Therefore, there is a need to investigate the effect of oxalic acid on the oxidation system. As shown in Fig. 3b, the addition of oxalic acid in the UV/PDS system significantly inhibited the decoloration of AY-17. The inhibition of oxalic acid has also been observed in other system. The UV–vis spectrum of 100 mmol/L oxalic acid solution (Fig. S2 in Supporting information) showed that oxalic acid has no absorption peak at 350-500 nm and therefore its photolysis under 365 nm UV light can be neglected. Ma et al. [58] reported that oxalic acid could complex with Fe3+ to generate Fe(Ⅲ)-oxalate complexes, which hindered the redox cycling of Fe3+/Fe2+ and led to the inhibition of ·OH formation and the decline of the degradation efficiency. However, in our UV/PDS system, it is impossible for oxalic acid to complex with Fe3+ and thus affect the formation of SO4·− radicals. It has also been reported that ·OH can decompose oxalic acid through electron transfer into carboxyl anion radicals (CO2·‒) (Eqs. 25 and 26), resulting in the consumption of active radicals and the loss of oxidative capacity [61, 62]. Similarly, oxalic acid might also compete with AY-17 for SO4·− through Eq. 27 [63], weakening the oxidation performance of the UV/PDS system. In addition, the generated CO2·− as a reductive radical is also likely to consume oxidizing substances (PDS, ·OH and SO4·−) in the UV/PDS system, thereby worsening the oxidation performance [63].
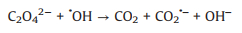
|
(25) |

|
(26) |
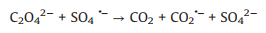
|
(27) |
For evaluating the effects of Cl− and Fe3+ ions on the mineralization degree of AY-17 during the photooxidative degradation, the TOC values of solutions before and after 90 min of irradiation were monitored, as presented in Fig. S3 (Supporting information). Although the decolorization rate of AY-17 reached 98.2% after 90 min, the mineralization degree was only about 6%. It can be concluded that the percentage of AY-17 degradation was much higher than the removal rate of TOC during the UV/PDS oxidation process. With the addition of 300 mmol/L Cl−, 23.5 mmol/L NO3‒ or 0.01 mmol/L Fe3+, the dye mineralization was accelerated, which is consistent with their promotion on dye degradation (Figs. 2a, 2d and 3a). Additionally, prolonging the UV irradiation time (7h) could lead to further mineralization and the TOC removal rate was up to 69.3%. The high decolorization rate and low mineralization rate indicated that partial decolorization of AY-17 in the UV/PDS system was possibly only due to the cleavage of the dye chromophores instead of the complete degradation.
AOX is an important parameter to evaluate the treatment performance of the contaminants degradation in the saline enviro nment. Herein, the formation of halogenated organic byproducts in the UV/PDS induced AY-17 degradation was quantitatively analyzed in the presence of 0 mmol/L and 300 mmol/L Cl−. As a chlorinated dye, the AOX of initial sample solution was 1.9 mg/L. In the absence of Cl−, the AOX level decreased to 1.5 mg/L after 90 min reaction due to the dechlorination of AY-17 during the UV/PDS oxidation. However, the AOX value did not decrease but had a few increase in a chloride-rich enviro nment (300 mmol/L). After 90 min reaction, the concentration of AOX in mixture solution was 2.1 mg/L. The increase of AOX value indicates the production and accumulation of more chlorinated compounds, and the similar effect of exogenic Cl− on AOX has also been observed in our previous studies on the degradation of chlorinated phenolic compounds [13, 16, 17, 19]. For example, Fang et al. [17] found that some new chlorinated compounds like 2, 4-dichlorophenol, 2, 4, 5-trichlorophenol and 2, 3, 5, 6-tetrachlorophenol were generated during the degradation of 2, 4, 6-trichlorophenol by the Co/PMS/Cl− system, as well as an increase about 20 mg/L in AOX value comparing to the Co/PMS system. The AOX accumulation was also observed in the UV/PDS system for monochlorophenols oxidation as the addition of Cl−, and accompanied with the formation and increase of toxic aromatic chlorinated compounds (such as dichlorophenols and 2, 3, 5, 3′, 5′-pentachloro-biphenyl) [13]. The reactive halogen species produced by exogenic X− can involve in the halogenation of organic substrates, and even low level of exogenic X− is able to cause high AOX accumulation, as reported by Sheng et al. [19].
Fig. S4 (Supporting information) shows the UV–vis spectra at 230−500 nm of AY-17 solution in the UV/PDS system versus reaction time. As the reaction proceeded, the characteristic absorption peaks at 402 nm continuously declined and almost disappeared with time, reflecting the destruction of the dye chromophore and conjugated system. Meanwhile, new peaks were observed in the wavelength range of 240−310 nm (Fig. S5 in Supporting information). Interestingly, the shoulder peak became more distinct when exogenic Cl− was added and continued to accumulate as the reaction proceeded. The phenomenon indicates that the addition of Cl− facilitated the formation of new byproducts such as chlorinated aromatic compounds. Repeta et al. reported that chlorinated aromatic acids showed strong absorption bands (240−400 nm) in the ultraviolet and near-ultraviolet regions [64]. Our previous studies also observed and confirmed the new absorbance peaks and the generation of several chlorinated compounds like 2, 4, 6-trichlorophenol, 1, 3, 5-trichloro-2-nitrobenzene and tetrachlorohydroquione in the presence of Cl− [7, 8].
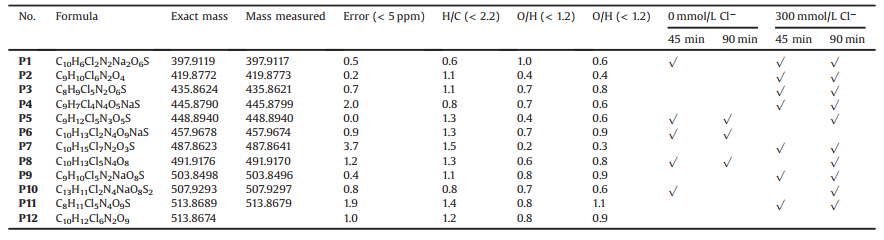
It was reported that the reaction intermediates formed during the degradation of AY-17 are more toxic than itself, and harm the respiratory and nervous systems of humans and organisms [21, 22]. It is difficult to determine the exact molecular structure by mass spectrometry alone, but the molecular composition of products can be determined more accurately by high resolution mass spectrometry. Therefore, the UPLC-Q-tof-MS analysis was performed to determine the main products generated in the photooxidation process of AY-17 in the absence and presence of 300 mmol/L Cl−. The pH value of the mixed solution before and after reaction was basically unchanged (pH 2.85–2.98), indicating that there was almost no generation of small molecular organic acids. Combined with Profinder software for data analysis, 12 possible degradation products were obtained (Table 1). In the absence of Cl−, 5 intermediates P1, P5, P6, P8 and P10 were detected in the reaction solution. As the reaction progressed, only P1 and P10 were completely degraded at 90 min, indicating that some refractory chlorinated compounds were still present in the solution. This provides direct evidences for the lower TOC removal (6%) and the still high AOX value (1.50 mg/L). When exogenic Cl− was added, other several polychlorinated (chlorine atom number > 3) organic intermediates (P2, P3, P4, P7, P9, P11 and P12) were identified. P1 still existed at 90 min andP5, P8 and P10 were only detected at 90 min. The addition of exogenic Cl− altered the degradation processes of organic compounds.
|
|
Table 1 Major conversion products of AY-17 detected by LC/MS-MS analysis in the UV/PDS/Cl− system. Experimental conditions: [AY-17]0 = 0.04 mmol/L, [PDS]0 = 10 mmol/L and pH 3.0. |
{kind=link}
{kind=link}
{kind=link}
The possible reaction mechanisms of AY-17 degradation are speculated: SO4·− radicals attacked the benzene ring in the organic molecule through electron transfer and mediated the formation of carbon-centered radical cations, which were then further oxidized to other organic byproducts and small molecules (CO2 and H2O) (Eq. 28) [3]. In addition, SO4·− radicals can also react with AY-17 and its byproducts through hydrogen extraction and the addition reactions on unsaturated bonds to achieve the cleavage of azo bond, pyrazole ring cleavage of the dye molecule, desulfonation or the substitution of chlorine atoms and other groups. The chlorine and sulfur atoms are partially mineralized and released as inorganic ions [32]. The decrease in AOX value in the absence of Cl‒ indicates the dechlorination during the oxidation of AY-17 by SO4·−. Several polychlorinated (chlorine atom number > 2) aromatic compounds (P5 and P8) were also detected, providing direct evidence for the dechlorination and subsequent rechlorination of the endogenic Cl‒ [20]. The degradation pathways of AY-17 involving Cl‒ might involve the one-electron oxidation of the dye radical by Cl2·−/Cl· and the generation of chlorinated byproducts (Eq. 29), where R is for the organic contaminants) [8, 13]. On the other hand, the chlorine element possesses strong electronegativity and Cl· readily occupies one electron to complete its octet [8]. Thus, another important pathway is related to the electrophilic substitution of the AY-17 and its byproducts initiated by Cl2·−/Cl· (Eq. 30), resulting in an undesirable increase in AOX in the UV/PDS system [10, 52].

|
(28) |
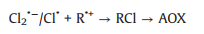
|
(29) |

|
(30) |
In Summary, the influencing parameters on AY-17 degradation under the UV/PDS oxidation process like substrate concentration, initial pH, inorganic ions (e.g., Cl−, and Br−), and metal ions were investigated. The acidic enviro nment and lower dye concentration are more favorable for the photooxidative degradation of AY-17. The pseudo-first order degradation rate constant is 1.56 × 10‒2 min‒1. The introduction of Cl−, Br−, Ag+ and Fe3+ could significantly improve the degradation efficiency of AY-17. In the UV/PDS system, the AOX value decreased with the degradation of AY-17, while that increased and accumulated in the presence of exogenic Cl−. The products identification by UPLC-Q-tof-MS confirms that more polychlorinated (chlorine atom number > 3) byproducts were formed in the UV/PDS/Cl ‒ system. The degradation reaction of AY-17 under the UV/PDS mainly involve the cleavage of azo bonds, the substitution of reactive radicals on the aromatic ring group and the cleavage of the pyrazole ring of the dye molecules. Notably, the introduction of exogenic Cl− accelerated the degradation and mineralization of AY-17, but also resulted in the production and accumulation of chlorinated organic compounds during oxidation reaction. Therefore, when evaluating the applicability of one oxidation process to the treatment of pollutants in saline dye wastewater, multiple comprehensive assessments such as kinetics, AOX, and degree of mineralization should be considered.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by National Key Research Development Program of China (No. 2019YFC0408304), the National Natural Science Foundation of China (No. 21677031), the Jiangsu Provincial Key Laboratory of Enviro nmental Science and Engineering (No. Zd1901) and Ningbo Natural Science Foundation (No. 202003N4135). Prof Weihua Song from Fudan University and Prof Wenhai Chu from Tongji University are appreciated for their help in LC–MS and AOX measurements, respectively.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.028.
| [1] |
R.S. Magazinovic, B.C. Nicholson, D.E. Mulcahy, D.E. Davey, Chemosphere 57 (2004) 329-335. DOI:10.1016/j.chemosphere.2004.04.056 |
| [2] |
K. Zhang, K.M. Parker, Environ. Sci. Technol. 52 (2018) 9579-9594. DOI:10.1021/acs.est.8b02219 |
| [3] |
G.P. Anipsitakis, D.D. Dionysiou, M.A. Gonzalez, Environ. Sci. Technol. 40 (2006) 1000-1007. DOI:10.1021/es050634b |
| [4] |
K.H. Chan, W. Chu, Water Res. 43 (2009) 2513-2521. DOI:10.1016/j.watres.2009.02.029 |
| [5] |
X.Y. Lou, Y.G. Guo, D.X. Xiao, et al., Environ. Sci. Pollut. Res. 20 (2013) 6317-6323. DOI:10.1007/s11356-013-1678-x |
| [6] |
Z. Wang, M. Feng, C. Fang, et al., RSC Adv. 7 (2017) 12318-12321. DOI:10.1039/C7RA01294B |
| [7] |
Z. Wang, R. Yuan, Y. Guo, L. Xu, J. Liu, J. Hazard. Mater. 190 (2011) 1083-1087. DOI:10.1016/j.jhazmat.2011.04.016 |
| [8] |
R. Yuan, S.N. Ramjaun, Z. Wang, J. Liu, J. Hazard. Mater. 196 (2011) 173-179. DOI:10.1016/j.jhazmat.2011.09.007 |
| [9] |
J. Zhou, J. Xiao, D. Xiao, et al., Chemosphere 134 (2015) 446-451. DOI:10.1016/j.chemosphere.2015.05.027 |
| [10] |
R. Yuan, S.N. Ramjaun, Z. Wang, J. Liu, Chem. Eng. J. 192 (2012) 171-178. DOI:10.1016/j.cej.2012.03.080 |
| [11] |
R. Yuan, Z. Wang, Y. Hu, B. Wang, S. Gao, Chemosphere 109 (2014) 106-112. DOI:10.1016/j.chemosphere.2014.03.007 |
| [12] |
J. Chen, Y. Qian, H. Liu, T. Huang, Environ. Sci. Pollut. Res. 23 (2016) 3824-3833. DOI:10.1007/s11356-015-5630-0 |
| [13] |
C. Fang, X. Lou, Y. Huang, et al., Chem. Eng. J. 323 (2017) 124-133. DOI:10.1016/j.cej.2017.04.094 |
| [14] |
G.D. Fang, D.D. Dionysiou, Y. Wang, S.R. Al-Abed, D.M. Zhou, J. Hazard. Mater. 227- 228 (2012) 394-401. DOI:10.1016/j.jhazmat.2012.05.074 |
| [15] |
J. Criquet, N.K.V. Leitner, Chemosphere 77 (2009) 194-200. DOI:10.1016/j.chemosphere.2009.07.040 |
| [16] |
C. Fang, Z. Wang, M. Feng, et al., Chemosphere 182 (2017) 624-629. DOI:10.1016/j.chemosphere.2017.05.065 |
| [17] |
C. Fang, D. Xiao, W. Liu, et al., Chemosphere 144 (2016) 2415-2420. DOI:10.1016/j.chemosphere.2015.11.030 |
| [18] |
C. Luo, J. Jiang, J. Ma, et al., Water Res. 96 (2016) 12-21. DOI:10.1016/j.watres.2016.03.039 |
| [19] |
B. Sheng, F. Yang, Y. Huang, et al., Chem. Eng. J. 381 (2020) 122634. DOI:10.1016/j.cej.2019.122634 |
| [20] |
L. Xu, R. Yuan, Y. Guo, et al., Chem. Eng. J. 217 (2013) 169-173. DOI:10.1016/j.cej.2012.11.112 |
| [21] |
J. Gao, Q. Zhang, K. Su, R. Chen, Y. Peng, J. Hazard. Mater. 174 (2010) 215-225. DOI:10.1016/j.jhazmat.2009.09.039 |
| [22] |
P.K. Malik, Dye. Pigment. 56 (2003) 239-249. DOI:10.1016/S0143-7208(02)00159-6 |
| [23] |
A. Huda, P.H. Suman, L.D.M. Torquato, et al., J. Photochem. Photobiol. A 376 (2019) 196-205. DOI:10.1016/j.jphotochem.2019.01.039 |
| [24] |
J. Khan, M. Tariq, M. Muhammad, et al., J. Chem. Chem. Eng. 39 (2018) 127-140. |
| [25] |
J. Khan, M. Tariq, M. Muhammad, et al., Chem. Soc. Ethiop. 33 (2019) 243-254. DOI:10.1155/2019/6192980 |
| [26] |
A. Khanna, V. Shetty, K. Environ, Sci. Pollut. Res. 20 (2013) 5692-5707. DOI:10.1007/s11356-013-1582-4 |
| [27] |
K.Y.A. Lin, T.-Y. Lin, Water Air Soil Pollut. 229 (2018) 10. DOI:10.1007/s11270-017-3648-2 |
| [28] |
Y. Huang, B. Sheng, Z. Wang, et al., Water Res. 145 (2018) 453-463. DOI:10.1016/j.watres.2018.08.055 |
| [29] |
E.E. Ebrahiem, M.N. Al-Maghrabi, A.R. Mobarki, Arab. J. Chem. 10 (2017) S1674-S1679. DOI:10.1016/j.arabjc.2013.06.012 |
| [30] |
O.S. Furman, A.L. Teel, M. Ahmad, M.C. Merker, R.J. Watts, J. Environ. Eng-Asce. 137 (2011) 241-247. DOI:10.1061/(ASCE)EE.1943-7870.0000323 |
| [31] |
C. Qi, X. Liu, J. Ma, et al., Chemosphere 151 (2016) 280-288. DOI:10.1016/j.chemosphere.2016.02.089 |
| [32] |
C.C. Liu, Y.H. Hsieh, P.F. Lai, C.H. Li, C.L. Kao, Dye. Pigment. 68 (2006) 191-195. DOI:10.1016/j.dyepig.2004.12.002 |
| [33] |
M.-S. Tsao, W.K. Wilmarth, J. Phys. Chem. 63 (1959) 346-353. DOI:10.1021/j150573a006 |
| [34] |
Y. Yang, J.J. Pignatello, J. Ma, W.A. Mitch, Environ. Sci. Technol. 48 (2014) 2344-2351. DOI:10.1021/es404118q |
| [35] |
P. Hu, M. Long, Appl. Catal. B: Environ. 181 (2016) 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| [36] |
P. Neta, R.E. Huie, A.B. Ross, J. Phys. Chem. Ref. Data 17 (1979) 1027-1284. DOI:10.1063/1.555854 |
| [37] |
S. Yang, P. Wang, X. Yang, et al., J. Hazard. Mater. 179 (2010) 552-558. DOI:10.1016/j.jhazmat.2010.03.039 |
| [38] |
Y.Q. Gao, N.Y. Gao, Y. Deng, Y.Q. Yang, Y. Ma, Chem. Eng. J.195- 196 (2012) 248-253. |
| [39] |
C. Liang, H.W. Su, Ind. Eng. Chem. Res. 48 (2009) 5558-5562. DOI:10.1021/ie9002848 |
| [40] |
X. Gu, S. Lu, L. Li, et al., Ind. Eng. Chem. Res. 50 (2011) 11029-11036. DOI:10.1021/ie201059x |
| [41] |
C. Liang, Z.S. Wang, C.J. Bruell, Chemosphere 66 (2007) 106-113. DOI:10.1016/j.chemosphere.2006.05.026 |
| [42] |
M.A. Rauf, S.S. Ashraf, Chem. Eng. J. 151 (2009) 10-18. DOI:10.1016/j.cej.2009.02.026 |
| [43] |
N. Daneshvar, M.A. Behnajady, M.K.A. Mohammadi, M.S.S. Dorraji, Desalination 230 (2008) 16-26. DOI:10.1016/j.desal.2007.11.012 |
| [44] |
G.P. Anipsitakis, D.D. Dionysiou, Environ. Sci. Technol. 38 (2004) 3705-3712. DOI:10.1021/es035121o |
| [45] |
V.K. Gupta, R. Jain, A. Nayak, S. Agarwal, M. Shrivastava, Mater Sci Eng. C 31 (2011) 1062-1067. DOI:10.1016/j.msec.2011.03.006 |
| [46] |
X.Y. Yu, Z.C. Bao, J.R. Barker, J. Phys. Chem. A 108 (2004) 295-308. DOI:10.1021/jp036211i |
| [47] |
F. Rehman, M. Sayed, J.A. Khan, et al., J. Hazard. Mater. 357 (2018) 506-514. DOI:10.1016/j.jhazmat.2018.06.012 |
| [48] |
Y. Ji, C. Dong, D. Kong, J. Lu, Q. Zhou, Chem. Eng. J. 263 (2015) 45-54. DOI:10.1016/j.cej.2014.10.097 |
| [49] |
L. Zhao, H. Hou, A. Fujii, M. Hosomi, F. Li, Environ. Sci. Pollut. Res. 21 (2014) 7457-7465. DOI:10.1007/s11356-014-2668-3 |
| [50] |
Y. Guo, J. Zhou, X. Lou, et al., Chem. Eng. J. 254 (2014) 538-544. DOI:10.1016/j.cej.2014.05.143 |
| [51] |
G.V. Buxton, C.L. Greenstock, W.P. Helman, A.B. Ross, J. Phys. Chem. Ref. Data 17 (1988) 513-886. DOI:10.1063/1.555805 |
| [52] |
J.E. Grebel, J.J. Pignatello, W.A. Mitch, Environ. Sci. Technol. 44 (2010) 6822-6828. DOI:10.1021/es1010225 |
| [53] |
P. Wang, S. Yang, L. Shan, R. Niu, X. Shao, J. Environ. Sci. 23 (2011) 1799-1807. DOI:10.1016/S1001-0742(10)60620-1 |
| [54] |
Y. Sun, J.J. Pignatello, Environ. Sci. Technol. 27 (1993) 304-310. DOI:10.1021/es00039a010 |
| [55] |
Z. Wang, J. Jiang, S. Pang, et al., Environ. Sci. Technol. 52 (2018) 11276-11284. DOI:10.1021/acs.est.8b02266 |
| [56] |
W. Peng, Y. Dong, Y. Fu, et al., Chem. Eng. J. 421 (2021) 127818. DOI:10.1016/j.cej.2020.127818 |
| [57] |
S. Garcia-Segura, E. Brillas, Water Res. 45 (2011) 2975-2984. DOI:10.1016/j.watres.2011.03.017 |
| [58] |
J. Ma, W. Ma, W. Song, et al., Environ. Sci. Technol. 40 (2006) 618-624. DOI:10.1021/es051657t |
| [59] |
A. Özcan, Y. Şahin, A.S. Koparal, M.A. Oturan, Water Res. 42 (2008) 2889-2898. DOI:10.1016/j.watres.2008.02.027 |
| [60] |
M. Pera-Titus, V. Garcı'a-Molina, M.A. Baños, J. Giménez, S. Esplugas, Appl. Catal. B: Environ. 47 (2004) 219-256. DOI:10.1016/j.jtice.2014.12.030 |
| [61] |
S. Gligorovski, R. Strekowski, S. Barbati, a.D. Vione, Chem. Rev. 115 (2015) 13051-13092. DOI:10.1021/cr500310b |
| [62] |
M. Vedrenne, R. Vasquez-Medrano, D. Prato-Garcia, et al., J. Hazard. Mater. 243 (2012) 292-301. DOI:10.1016/j.jhazmat.2012.10.032 |
| [63] |
Z. Wang, W. Qiu, S. Pang, J. Jiang, Water Res. 164 (2019) 114957. DOI:10.1016/j.watres.2019.114957 |
| [64] |
D.J. Repeta, N.T. Hartman, S. John, A.D. Jones, R. Goericke, Environ. Sci. Technol. 38 (2004) 5373-5378. DOI:10.1021/es049921q |