 2021, Vol. 32
2021, Vol. 32 


b Shanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China;
c Institute of Functional Materials, Donghua University, Shanghai 201620, China
Antimony (Sb) compounds are emerging contaminants of global concern [1, 2]. The presence of Sb in water bodies has been routinely reported due to release from diverse sources. The US Environmental Protection Agency has classified Sb compounds as priority pollutants. Because of their high toxicity, a maximum level of 6 μg/L has been established for Sb compounds in drinking water [3].
Sb has similar chemical properties to arsenic (As) and both are in the same group (VA) of the periodic table. However, compared with As, only limited efforts have been devoted to the removal of Sb from contaminated water [4]. The similarities in chemical properties are often emphasized with the aim of transferring existing removal methods for As to Sb. However, the physical properties of Sb and As are dissimilar. For example, Sb has a relatively large atomic radius (Sb = 1.41 Å vs. As = 1.21 Å) and slower mobility in solution compared with As [5]. Thus, the transfer of removal methods used for As, to Sb, may be inappropriate. In addition, the predominant inorganic Sb species under anoxic conditions is antimonite (Sb(III)), which is 10 times more toxic than antimonate (Sb(V)) [3]. Sb(III) is also neutrally-charged over a wide pH range (from 2 to 10.4) [6], and is difficult to remove due to its high mobility and weak affinity towards sorbents. Therefore, oxidative transformation of Sb(III) to less toxic Sb(V) (see Eq. 1) has been regarded as a viable and effective pre-treatment approach.

|
(1) |
This strategy not only achieves detoxification of Sb(III), but also facilitates post-treatment efficacy as Sb(V) is negatively-charged and less toxic. To this end, hydrogen peroxide (H2O2) is a plausible, moderate and benign oxidant (E = 1.54 V vs. Ag/AgCl). However, based on our preliminary results, the effective oxidation of Sb(III) by H2O2 required strong alkaline conditions.
To further enhance the oxidizing power of H2O2, processes employing transition metal (e.g., Fe, Cu, Mn and Ce) oxide catalysts to activate H2O2 have been extensively pursued [7]. These Fenton-like processes proceed via radical and non-radical pathways. The radical mechanism involves the generation of hydroxyl radicals (∙OH) and other reactive oxygen species via cleavage of the peroxide bond of H2O2 by catalysts [8]. In this instance, H2O2 assumed the role of radical precursor and high concentrations of H2O2 were usually required to overcome radical quenching. In contrast, the non-radical pathway involved the formation of metal-hydroperoxo surface complexes [9]. The non-radical mechanism has attracted much attention because of its high selectivity towards target compounds of concern. For example, Shan et al. [10] reported that Ce-hydroperoxo surface complexes were responsible for the selective oxidation of As(III) in solution, while dissolved O2, H2O2, and ∙OH made negligible contributions to the process. Despite advances in the understanding of the non-radical mechanism, some practical limitations remain: i) Sluggish reaction kinetics; ii) self-aggregation of nanoscale catalysts; iii) hazards associated with H2O2 usage; and iv) generation of Fe sludge when using Fe-based catalysts.
Previous investigations were mainly performed in conventional batch reactors, in which the kinetics were typically mass transfer limited due to the dependence on slow diffusion [11]. For example, at [H2O2]0/[As(III)]0 of 5.0, a complete catalytic conversion of 35 mg/L As(III) to As(V) by H2O2 over cerium oxide nanoparticles could only be obtained after a 4 h reaction [10]. In contrast, mass transport in a flow-through configuration is predominantly controlled by convection, which can significantly increase the overall reaction kinetics [12]. For example, compared with conventional batch electrochemistry, tetracycline removal kinetics and degradation efficiency showed increases of approximately three- and two-fold, respectively, using a flow-through configuration [13]. In addition, to overcome agglomeration issues, nanoscale catalysts are often supported on a porous solid with a high specific surface area. Among these support materials, the one-dimensional carbon nanotubes (CNTs) have attracted attention due to their combined mechanical, physicochemical and electric characteristics [14, 15]. The ability to form one-dimensional CNT structures into stable, high surface area, and conductive networks would permit dual functionality as both electrodes and filtration media [16]. Another benefit of CNT is their potential to generate H2O2 in situ by catalyzing the two-electron reduction of O2 in aqueous solution using an electric field [13]. Such a strategy would also eliminate the potential hazards associated with transport, storage and usage of H2O2.
Both TiO2 and CeO2 were shown to be effective nonferrous Fenton-like catalysts [10,17], while a Ti-Ce binary oxide exhibited enhanced catalytic activity [9]. Previously we also demonstrated the synergistic effect of a Ti-Ce binary oxide for phosphite removal [18]. Here, we proposed a flow-through electro-Fenton-like system for the efficient transformation of highly toxic Sb(III) to Sb(V) via a non-radical pathway. The key to this system was an anodic CNT filter functionalized with Ti-Ce binary oxide nanoparticles. Enhanced electro-Fenton performance was expected due to the synergistic effects of flow-through configuration, in situ generated H2O2, improved mass transport, abundant exposed active sites, and Ti-Ce binary oxide catalyst. The underlying mechanisms were investigated and discussed in detail. We hypothesized: i) Dissolved O2 in the Sb(III) solution would be reduced to H2O2 under an electric field; ii) H2O2 would react with Ti-Ce binary oxide to form hydroperoxo complexes for the efficient oxidation of Sb(III) via a non-radical pathway; iii) the flow-through design would accelerate the conversion of Sb(III) to Sb(V) by convection. To the best of our knowledge, the transformation of Sb(III) to Sb(V) via non-radical pathway and the underlying mechanisms have not been reported elsewhere.
The Ti/Ce binary oxide functionalized CNT (Ti/CeOx-CNT) filters were prepared by a co-precipitation procedure with modifications [19]. Details on the preparation and characterization of Ti/CeOx-CNT hybrid filters, and experimental details were available in Supporting information.
Fig. S2 (Supporting information) shows the X-ray diffraction (XRD) patterns of CNT, TiO2-CNT, CeO2-CNT, and Ti/CeOx-CNT (with varying Ti/Ce ratios) filters. Crystal structures of TiO2 were absent in the TiO2-CNT filter either due to the limited TiO2 loading or the presence of amorphous TiO2. The XRD pattern for CeO2-CNT was characteristic of cerianite (CeO2, JCPDS No. 34-0394). A similar pattern was also obtained for the Ti/CeOx-CNT filter, indicating that doping with Ti had not destroyed the crystal phase of CeO2. As the doping of Ti into CeO2 increased, the crystal peaks of the cerianite phase gradually weakened. This could be explained by the differences in atomic radii between Ce and Ti (Ce4+ = 0.97 Å; Ce3+ = 1.14 Å; and Ti4+ = 0.61 Å) [20]. Compared to CeO2-CNT filter, the cerianite peak signals of the Ti/CeOx-CNT filters were obviously weakened. No evident anatase characteristic peaks were identified as well indicating the smaller atomic radius Ti was doped into CeO2 lattice, and led to the reduction of Ce-O-Ce symmetric structure and the appearance of Ce-O-Ti bond. The characteristic broadened peaks in the XRD patterns from the CeO2-CNT and Ti/CeOx-CNT filters revealed that those loaded nanoscale particles were 6.3 nm and 3.8 nm, respectively, according to the Debye-Scherrer formula [10]. Compared with the pristine CNT filter (Fig. S3a in Supporting information), field emission scanning electron microscopy (FESEM) characterization showed that the Ti/CeOx-CNT filter had a rough surface with an increased diameter of 25 ± 8 nm, due to the deposition of Ti/CeOx nanoparticles (Fig. S3b in Supporting information). Furthermore, the visualized energy-dispersive spectroscopy mapping data of the selected region of the Ti/CeOx-CNT filter (Ti:Ce=2:1) showed an uniform distribution of C, O, Ti and Ce on the CNT walls (Figs. S3c–f and S4a in Supporting information). Additionally, Ti/CeOx and CeO2 lattice fringes of similar spacing (0.30 nm) were observed (Fig. S4b in Supporting information). This suggested the possible intercalation of Ti doped into the cerianite lattice to replace Ce atoms during the co-precipitation step. It also implied that Ti in the amorphous phase binds with Ce through O (Ce-O-Ti), while the remaining Ce forms the Ce-O-Ce structure [19]. The TGA results suggested that the loading of Ti/CeOx onto an effective Ti/CeOx-CNT filtration area of 7.1 cm2 was 58.4wt% (Fig. S5a in Supporting information). Since Ti(SO4)2 is easy be hydrolyzed, the loading of Ti onto the hybrid would be rather limited. This hypothesis was further verified by the ICP-MS analysis, which indicated that 0.11 g/g Ti and 0.38 g/g Ce were present on the Ti:Ce=2:1 filter. As displayed in Fig. S5b (Supporting information), linear sweep voltammetry (LSV) tests suggested that the Ti:Ce=2:1 filter exhibit an evidently improved hydrogen evolution reaction activity as compared with the Ti:Ce=1:1 and Ti:Ce=1:2 filters. At a current density of 10 mA/cm2, the potential for the Ti:Ce=2:1 filter (1.232 V) is 271 mV and 201 mV more positive than that of Ti:Ce=1:1 (1.503 V) and Ti:Ce=1:2 (1.433 V) filter, respectively.
Fig. S6 (Supporting information) shows that the superficial elemental ratios obtained by X-ray photoelectron spectroscopy (XPS) analysis were 97% C and 3% O for the CNT and 88.4% C, 9.7% O, 1.4% Ce and 0.5% Ti for the Ti/CeOx-CNT filter. Fig. S7 (Supporting information) shows the XPS of the Ce 3d from the Ti/CeOx-CNT filter. The results suggested that the ratio of Ce3+ increased as the Ti loading increased from 23.3% (Ti:Ce=1:2) to 34.8% (Ti:Ce=2:1). Similar observations were also reported in recent studies [21, 22]. According to previous reports [21, 23], the bands labeled as ii and ii' in Fig. S7 were ascribed to Ce3+ and while those labeled as i, iii, iv, i', iii' and iv' were assigned to Ce4+. This phenomenon indicated that Ce3+ and Ce4+ coexist on the surface of the Ti/CeOx-CNT filter, and Ce4+ was the dominant species. The partial reduction of Ce4+ could be attributed to the incorporation of Ti into Ce-O-Ce to form Ce-O-Ti microstructures. Collectively, the results were consistent with the successfully preparation of the Ti/CeOx-CNT filter.
A flow-through Fenton-like system was established using the prepared Ti/CeOx-CNT filter as a functional cathode and a perforated Ti foil as the anode. Sb(III) was selected as the probe compound to evaluate system efficacy for the transformation of Sb(III) to Sb(V). H2O2 could be in situ produced via a 2-electron oxygen reduction reaction at the Ti/CeOx-CNT filter cathode (Eq. 2). The H2O2 yield as a function of applied potential using a CNT network as cathode is shown in Fig. S8 (Supporting information). The influent stream containing 10 mmol/L Na2SO4 as the electrolyte was pre-sparged with oxygen for 30 min at pH 6.5. The results showed that as the applied cathode potential was decreased from ‒0.2 V to ‒0.6 V, the corresponding H2O2 flux increased from 2.56 mmol h−1m−2 to 4.29 mmol h−1m−2. This was consistent with the occurrence of the 2-electron oxygen reduction reaction. However, a further decrease in the applied cathode potential to ‒0.8 V reduced the H2O2 flux to 2.18 mmol h−1m−2, possibly due to the emergence of competing reactions, such as H2 production and reduction of H2O2 to H2O [24].

|
(2) |
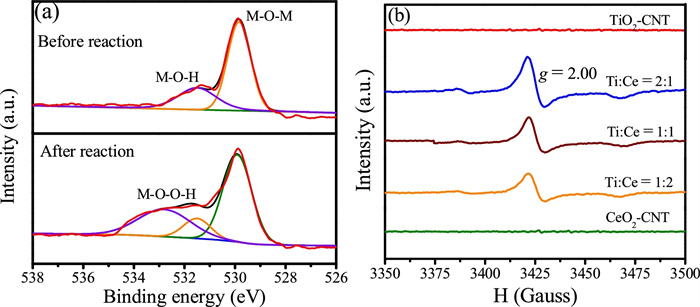
To eliminate the contribution of physical adsorption by the Ti/CeOx-CNT filter on Sb(III) removal, Sb(III) was adsorption saturated on the hybrid filters prior to application of the electric field. The effects of operational parameters on the kinetics of the Fenton-like oxidation of Sb(III) including Ti/CeOx loading level, solution pH, applied voltage, and flow rate were systematically investigated. Fig. S9a (Supporting information) shows the effect of Ti:Ce ratios on the oxidation of Sb(III). As expected, the pristine CNT filter demonstrated relative low Sb(III) conversion efficiency (38%) at ‒0.4 V and neutral pH, while and the TiO2-CNT and CeO2-CNT filters gave slightly increased oxidation efficiencies of 47% and 42%, respectively, under similar conditions. In contrast, the Ti-Ce binary oxide co-modified CNT filter demonstrated a much enhanced Sb(III) conversion efficiency. This enhanced catalytic activity toward Sb(III) oxidation could be attributed to their intrinsically favorable micro-structures. Similar bimetallic oxides have shown excellent selectivity and superior oxidation performance when compared with their mono-oxide counterparts because of the synergistic effects of the binary catalyst [25]. It also has been reported that oxygen vacancies play an important role in determining the electrocatalytic activity. The incorporation of Ti into CeO2 may boost the storage capacity of mobile oxygen by CeO2. Fig. 1a shows the O 1s XPS spectra of the Ti/CeOx-CNT filter (Ti:Ce=2:1) before and after adsorption of Sb(III). The peak centered at 529.8 eV could be due to the lattice oxygen while a new peak appearing at 531.5 eV might be assigned to oxygen vacancies [9]. Other fingerprint evidence for probing oxygen vacancies could be observed using electron paramagnetic resonance (EPR) spectroscopy, as shown in Fig. 1b. The typical EPR signal at g=2.00 obtained for the Ti/CeOx-CNT filter indicated the existence of isolated oxygen vacancies within the binary oxide [26], while no peaks were identified for the TiO2 or CeO2-functionalized filters. It has been reported that oxygen vacancies, which are pervasive in metal oxides, could facilitate the charge transport kinetics, thus improving electrocatalytic performance [27].

|
Download:
|
| Fig. 1. (a) XPS O 1s spectra of the Ti/CeOx-CNT filter (Ti:Ce=2:1) before and after Sb(III) adsorption (M stands Ti or Ce). (b) EPR spectra of TiO2, CeO2, and Ti/CeOx powders. | |
{kind=link}
The Ti:Ce ratio was also important in determining the performance of the Fenton-like system. For example, at Ti:Ce ratio of 2:1, a Sb(III) conversion of > 99% was observed with a single-pass through the filtration system. Under the same conditions, the conversion of Sb(III) at Ti:Ce ratios of 1:1 and 1:2 were 77.4% and 57.6%, respectively. This phenomenon could be partially explained by the size effect of the nano-catalysts [28]. Based on the XRD results and Debye-Scherrer formula, the dimensions of the Ti-Ce binary oxide increased from 3.8 nm to 6.3 nm as the Ti content decreased from 20 mmol/L to 0 mmol/L (Table S1 in Supporting information). Under these conditions, the corresponding Sb(III) oxidation efficiency reduced from > 99% to 42%. The highest catalytic activity was observed at a Ti:Ce ratio of 2:1 which represented the smallest crystal size of 3.8 nm. This relationship between the crystal size and the corresponding Sb(III) oxidation efficiency showed a strong linear dependence (R2 > 0.99).
The applied electric field can directly influence the H2O2 yield, thereby affecting kinetics of the Sb(III) detoxification [13]. Fig. S9b (Supporting information) shows that the oxidation efficiency of Sb(III) increased as cathode potential decreased from ‒0.2 V to ‒0.4 V. A maximum Sb(III) oxidation efficiency of 99.1% was obtained between ‒0.4 V and ‒0.6 V, and this was consistent with the increasing yield of H2O2 in this potential range. As the applied potential was decreased further (‒0.8 V), the yield of H2O2 decreased, probably due to competing side reactions (Eqs.3-5). The flow rate of the system and the pH of the solution were also explored in Supporting information.

|
(3) |

|
(4) |

|
(5) |
Previous studies have proposed a catalytic oxidation mechanism based on the formation of interfacial peroxide species in the presence of CeO2/TiO2 and H2O2 (Eq. 6) [9, 17]. EPR, radical scavenging, and other control experiments were used to determine whether ∙OH (conventional Fenton systems) or Ti/Ce-hydroperoxo surface complexes (Eqs. 7 and 8) contributed to the effective Sb(III) transformation.

|
(6) |

|
(7) |

|
(8) |
where M stands for a metal atom (Ce or Ti).
Fig. 2a shows the EPR spectra obtained using 5, 5-dimethyl-1-pyrroline n-oxide (DMPO) as the trapping agent for ∙OH. No characteristic peaks were observed regardless of the applied potentials, suggesting that a non-radical mechanism may dominate the Sb(III) conversion process. This was further supported by radical scavenging experiments using TBA (kOH∙ = 3.0 × 109 L mol−1 s−1) added at different concentrations (Fig. S10 in Supporting information). The contribution of ∙OH to the Sb(III) oxidation process was negligible. To eliminate the influence of H2O2, a solution of Sb(III) (200 mL of 500 μg/L) with Na2SO4 electrolyte (10 mmol/L) was pre-purged with saturated nitrogen. As expected, the Sb(III) transformation efficiency declined significantly from 99.1% to 25.7% (Fig. 2b), indicating that the formation of the interfacial hydroperoxo complexes (≡TiOOH and ≡CeOOH) had not occurred for Sb(III) oxidation to proceed (Eqs. 3 and 5). To confirm the key role of the hydroperoxo surface complexes, the Ti/CeOx-CNT filter presumably the generation of H2O2 was completely inhibited, was pretreated with H2O2 prior to contact with Sb(III). Fig. 2c shows that the H2O2-pretreated filter could maintain Sb(III) conversion (79.5%) without additional H2O2. This result demonstrated that the oxidation of Sb(III) over the binary oxide catalyst was mainly mediated by the hydroperoxo surface complexes and the non-radical nature of the interfacial complex. Inspection of the O 1s XPS spectrum obtained from Ti/CeOx-CNT filter with a Ti:Ce ratio of 2:1 (Fig. 1a) shows that an extra peak at 532.8 eV appeared after reaction, attributed to oxygen (‒1 valance state) in the surface complexed peroxide species (M-OOH). These results provided solid evidences for the formation of surface hydroperoxo complexes which act as the dominant oxidizing species in the electro-Fenton-like oxidation of Sb(III) over the Ti/CeOx-CNT filter. The overall objective was to clarify the oxidizing active species and the possible mechanism within the proposed Fenton-like system (Fig. 3).

|
Download:
|
| Fig. 2. Identification of dominant oxidizing species in the electro-Fenton-like system: (a) EPR spectra obtained with the ∙OH trapping agent DMPO at various applied potentials. (b) Comparison of the Sb(III) conversion kinetics under saturated N2 and O2 conditions. (c) Comparison of the Sb(III) conversion kinetics using H2O2-pretreated Ti/CeOx-CNT filter at 0 V and using Ti/CeOx-CNT filter at ‒0.4 V. | |
{kind=link}

|
Download:
|
| Fig. 3. Proposed Sb(III) oxidation mechanism using the flow-through electro-Fenton-like system. | |
{kind=link}
A novel flow-through electro-Fenton-like system was designed and evaluated for the efficient detoxification of aqueous Sb(III) via non-radical pathways. The combination of sufficient exposed active sites, flow-through design, and electroactivity of the filter, enabled an effective and robust detoxification of aqueous Sb(III). This study has demonstrated a robust and effective strategy for the detoxification of water containing Sb(III) and other similar heavy metal ions by integrating state-of-the-art advanced oxidation processes, electrochemistry and nano-filtration technology.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThis work was supported by the Natural Science Foundation of Shanghai, China (No. 18ZR1401000).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.02.007.
| [1] |
B. Dousova, M. Lhotka, F. Buzek, et al., Sci. Total Environ. 702 (2020) 134642. DOI:10.1016/j.scitotenv.2019.134642 |
| [2] |
X.J. Guo, Z.J. Wu, M.C. He, Water Res. 43 (2009) 4327-4335. DOI:10.1016/j.watres.2009.06.033 |
| [3] |
Z.J. Wu, M.C. He, X.J. Guo, R.J. Zhou, Sep. Purif. Technol. 76 (2010) 184-190. DOI:10.1016/j.seppur.2010.10.006 |
| [4] |
M.N. Zhang, J. Jia, K. Huang, X.D. Hou, C.B. Zheng, Chin. Chem. Lett. 29 (2018) 456-460. DOI:10.1016/j.cclet.2017.09.062 |
| [5] |
Z.P. Fu, G.P. Zhang, H.X. Li, et al., Int. J. Soil Sediment Water 16 (2016) 2471-2481. DOI:10.1007/s11368-016-1484-4 |
| [6] |
G. Ungureanu, S. Santos, R. Boaventura, C. Botelho, J. Environ. Manage. 151 (2015) 326-342. DOI:10.1016/j.jenvman.2014.12.051 |
| [7] |
C. Wang, Y. Liu, T. Zhou, et al., Chin. Chem. Lett. 30 (2019) 2231-2235. DOI:10.1016/j.cclet.2019.08.055 |
| [8] |
J. Wang, Z.X. Wu, L.L. Han, et al., Chin. Chem. Lett. 27 (2016) 597-601. DOI:10.1016/j.cclet.2016.03.011 |
| [9] |
C. Shan, H. Liu, M. Hua, B.C. Pan, Environ. Sci. Technol. 54 (2020) 5893-5901. DOI:10.1021/acs.est.0c00159 |
| [10] |
C. Shan, Y. Liu, Y.H. Huang, B.C. Pan, Environ. Int. 124 (2019) 393-399. DOI:10.1016/j.envint.2019.01.014 |
| [11] |
T. Noel, Y.R. Cao, G. Laudadio, Acc. Chem. Res. 52 (2019) 2858-2869. DOI:10.1021/acs.accounts.9b00412 |
| [12] |
G.D. Gao, Q.Y. Zhang, Z.W. Hao, C.D. Vecitis, Environ. Sci. Technol. 49 (2015) 2375-2383. DOI:10.1021/es505679e |
| [13] |
F.Q. Liu, Y.B. Liu, Q.F. Yao, et al., Environ. Sci. Technol. 54 (2020) 5913-5921. DOI:10.1021/acs.est.0c00427 |
| [14] |
M.F.L. De Volder, S.H. Tawfick, R.H. Baughman, A.J. Hart, Science 339 (2013) 535-539. DOI:10.1126/science.1222453 |
| [15] |
Y.B. Liu, G.D. Gao, C.D. Vecitis, Acc. Chem. Res. (2020) 2892-2902. DOI:10.1021/acs.accounts.0c00544 |
| [16] |
C.D. Vecitis, G.D. Gao, H. Liu, J. Phys. Chem. C 115 (2011) 3621-3629. DOI:10.1021/jp111844j |
| [17] |
D.H. Kim, A.D. Bokare, M. Koo, W. Choi, Environ. Sci. Technol. 49 (2015) 3506-3513. DOI:10.1021/es5056897 |
| [18] |
F.Q. Liu, Y.B. Liu, C.S. Shen, et al., Sci. Total Environ. 710 (2020) 135514. DOI:10.1016/j.scitotenv.2019.135514 |
| [19] |
P. Li, Y. Xin, Q. Li, et al., Environ. Sci. Technol. 46 (2012) 9600-9605. DOI:10.1021/es301661r |
| [20] |
Z.N. Shi, P. Yang, F. Tao, R.X. Zhou, Chem. Eng. J. 295 (2016) 99-108. DOI:10.1016/j.cej.2016.03.032 |
| [21] |
W.R. Zhao, Y. Tang, Y.P. Wan, et al., J. Hazard. Mater. 278 (2014) 350-359. DOI:10.1016/j.jhazmat.2014.05.071 |
| [22] |
Z.W. Fu, Y.Y. Zhong, Y.H. Yu, et al., ACS Omega 3 (2018) 198-207. DOI:10.1021/acsomega.7b01475 |
| [23] |
F.Y. Gao, X.L. Tang, H.H. Yi, et al., Chem. Eng. J. 317 (2017) 20-31. DOI:10.1016/j.cej.2017.02.042 |
| [24] |
H. Liu, A. Vajpayee, C.D. Vecitis, ACS Appl. Mater. Inter. 5 (2013) 10054-10066. DOI:10.1021/am402621v |
| [25] |
Z.B. Liang, C. Qu, W.Y. Zhou, et al., Adv. Sci. 6 (2019) 1802005. DOI:10.1002/advs.201802005 |
| [26] |
Y.C. Huang, H.X. Hu, S.X. Wang, et al., Appl. Catal. B: Environ. 218 (2017) 700-708. DOI:10.1016/j.apcatb.2017.07.028 |
| [27] |
H.S. Kim, J.B. Cook, H. Lin, et al., Nat. Mater. 16 (2016) 454-460. DOI:10.1038/nmat4810 |
| [28] |
B.C. Hodges, E.L. Cates, J.H. Kim, Nat. Nanotechnol. 13 (2018) 642-650. DOI:10.1038/s41565-018-0216-x |