 2021, Vol. 32
2021, Vol. 32 



b Institute for Ecological Research and Pollution Control of Plateau Lakes, School of Ecology and Environmental Science, Yunnan University, Kunming 650504, China;
c Department of Physics, Umeå University, Umeå 901 87, Sweden
Various kinds of dyes have been widely applied in many industries, including textile, pulp and paper, pharmaceutical, and food manufacturing industries [1, 2]. Efficient and thorough treatment has a profound impact on industrial development [3, 4]. Considerable researches [5-7] have demonstrated that advanced oxidation processes (AOPs) can efficiently degrade organic pollutants owing to the reactive oxygen species with strong oxidation capacity. The main AOPs are based on hydroxide (·OH) and sulfate radicals (SO4·−) [8], among which Fenton oxidation technology is the most mature. ·OH radicals are challenging, as their use includes explosive reagents (H2O2), narrow pH, short life time of free radicals (< 1 μs), and secondary pollution [9-13]. Compared with ·OH (E0=2.8 eV) [14, 15] produced by H2O2, sulfate radicals (SO4·−) have attracted increasing attention [16-18] due to their high oxidation potential (E0=2.5–3.1 eV), longer lifetime (30–40 μs) [19-21], and wider pH range [3, 22, 23]. The sources of sulfate radicals are peroxysulfate (PS and PMS). Simultaneously, PMS was found to be ecologically friendly, water solubility, safe and high stability [24].
Furthermore, there are many approaches to activate PS or PMS to produce SO4·−, including heat [25-27], base [28], UV light [25, 26], ultrasound [27, 28], metal-free catalysts [18, 29], and metal catalysts [30-33]. Co-based catalysts have been proven to be the most efficient materials for activating PMS in organic pollutant removal [34, 35]. It should be noted that homogeneous Co(Ⅱ) shows efficient activity for mineralizing pollutants because the active sites can be fully utilized. However, Co(Ⅱ) cannot be recovered after the reaction, and has been considered toxic and harmful to human health, which limits its scalability. The second type of Co-based catalyst is a single-atom catalyst. Huang et al. [39] developed a CFs-CoPc/PMS/HCO3− system to decolorize 50 μmol/L acid red 1 (AR1), with almost 100% decolorization efficiency in 35 min. However, the energy-consuming preparation process, high cost, and low content of Co remain challenging. Compared with the above two kinds of catalysts, bimetallic Co-based oxides have received attention because of their high catalytic efficiency, large specific surface area, adjustable structure, and magnetic separation. Yang et al. [24] prepared CoFe2O4-SAC nanocomposites to decontaminate norfloxacin by activating PMS. Thereafter, Yao et al. [36] synthesized a series of CoxMn3-xO4 particles as PMS catalysts.
Among the bimetallic Co-based oxides, NiCo2O4 possesses multiple convertible valence states, fast electron transfer, high stability and safety, and controllable morphology. Note that Co and Ni, as transition metals, can change their valence states simultaneously, resulting in notable catalytic activity. However, researchers have only focused on the electrochemical properties of NiCo2O4 [37, 38]. Catalytic properties have seldom been studied, which may be due to the restrictions stemming from the difficulty in controlling morphologies and dispersities. To control the morphology and dispersity, Zhang et al. [39] synthesized porous NiCo2O4 nanoflakes with the assistance of DMF and EDTA to activate PMS. Tian et al. [40] successfully prepared dandelion-like NiCo2O4 microspheres with the help of transition metals and byproducts from the hydrolysis of urea.
Inspired by the preparation method of Tian et al. [40], in this study, we used NH3·H2O based on the reaction of Co2+/Ni2+ with NH3/OH−, further enhancing the nucleation of NH3 vesicles from the hydrolysis of NH3·H2O to prepare an open-type NiCo2O4 hollow-sphere (Scheme S1 in Supporting information), which has not yet been studied. This method is simple, and the process of removing the template was omitted. Furthermore, the hole on the surface of the synthesized catalyst can minimize the diffusion resistance of reactants and products, which can allow PMS to enter the inner chamber; further, the inner and outer surfaces of the open-type NiCo2O4 hollow-sphere were utilized simultaneously, enhancing the performance of the PMS. The effects of various operational parameters were also studied. TOC removal rate can reach up to 80% in NiCo2O4/PMS system. Finally, the possible activation mechanism was further discussed based on radical quenching studies and the electron spin response (ESR) technique.
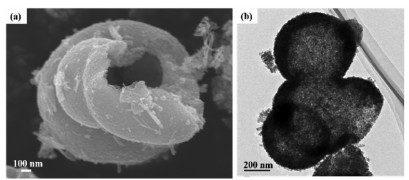
Typical SEM images in Fig. 1a revealed well-defined, open-type hollow spheres for each individual unit. The inner surface was rough and uneven because the specific open-type hollow-sphere was composed of numerous small-sized nanoparticles, which provide a larger contact area than the smooth surface. The TEM image in Fig. 1b showed that the diameter of hollow spheres was approximately 500 nm. Each hollow-sphere had a hole on the surface, which minimized the diffusional resistance in the system and enabled sulfate radicals and rhodamine B (RhB) to diffuse freely into the interior. After degradation, the products spreaded into the exterior due to concentration differences, indicating that the reaction occurred simultaneously on the inner and outer surfaces, and the contact area with the reactants was doubled. Besides the basic morphology properties, X-ray diffractometer (XRD) patterns, N2 physical adsorption apparatus, X-ray photoelectron spectroscopy (XPS) were provided in Figs. S1 and S2 (Supporting information). The results of all characterization techniques showed that NiCo2O4 has a suitable morphology, large specific area, and pollutants. The effects of operation parameters like PMS dose, NiCo2O4 dose, initial pH, ambient temperature and coexisting ions and the rate constant (k) of the NiCo2O4/PMS system were shown in Figs. S3 and S4 (Supporting information) together with cycle experiments shown in Fig. S5a (Supporting information). Meanwhile, as manifested in Fig. S5b (Supporting information), this catalyst possesses the property of magnetic separation, which simplifies the recycling process. After five cycles, the removal efficiency remained at 90%. The decrease in the removal efficiency might be due to the loss of the reduction of Co2+, from the XPS characterization.

|
Download:
|
| Fig. 1. SEM(a) and TEM(b) of NiCo2O4. | |
{kind=link}
Fig. 2a showed the color change over time. Within 40 min, the color faded. As shown in Fig. 2b, the final removal rate reached 100% within 40 min, while the removal efficiency of TOC was 80%, which was much more than that of the commercial catalyst about 20%.

|
Download:
|
| Fig. 2. (a) Degradation process, and (b) comparison of removal of RhB and TOC. Experimental conditions: [RhB]=100 mg/L, NiCo2O4 dose=133.33 mg/L, PMS dose=166.67 mg/L, pH 4.81, T = 25 ℃. | |
{kind=link}
Based on the above results, to highlight the advantages of this reaction system, the reaction systems reported in literature were compared with those of NiCo2O4/PMS. The results are shown in Table S1 (Supporting information). When the oxidizing agent was H2O2, the reaction rate constant (k) was lower, which can be explained by the fact that the oxidation capacity of ·OH was weaker than that of SO4·−. When the oxidizing agent was PMS, although the activation energy was larger or smaller than that in this study, the reaction conditions in the study were more economic and milder. Note that the NiCo2O4/PMS system can handle highly concentrated organic pollutants with a superior removal rate.
In general, active radicals are responsible for the oxidation of dye in PMS activation technology. tert-Butyl alcohol (TBA) without α-H as a chemical probe for ·OH (3.8–7.6 × 108 L mol−1 s−1) and methanol (MeOH) with α-H as a chemical probe for SO4·− (1.6–7.7 × 107 L mol−1 s−1) and ·OH (1.2–2.8 × 109 L mol−1 s−1) were used to identify the active radicals [24, 41]. As presented in Fig. S6a (Supporting information), the inhibition was more obvious with the increase in TBA and MeOH. The suppressive effect was more significant in the MeOH-PMS system. From these results, it can be concluded that both SO4·− and ·OH were present in this reaction, but the contribution of SO4·− was greater in the NiCo2O4/PMS system. However, when the initial pH was adjusted to 11.00 (Fig. S6b in Supporting information), the role of ·OH was more obvious.
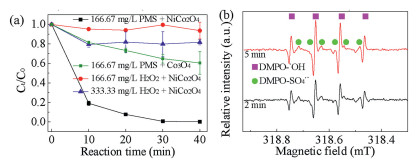
To validate the above speculation, the ESR technique was used to detect the active radicals in the NiCo2O4/PMS system (Fig. 3a). The signals of DMPO-·OH and DMPO-SO4·− both exist in the ESR spectrum, showing that both SO4·− and ·OH could be produced in the NiCo2O4/PMS system for RhB degradation. As shown in Fig. 3b, NiCo2O4 was used to catalyze H2O2 and PMS, and the removal efficiency in the NiCo2O4/H2O2 system was lower than that in the NiCo2O4/PMS system, even when the concentration of H2O2 increased. This proved that NiCo2O4 is the exclusive catalyst for activating PMS. Moreover, under the same conditions, the activity of NiCo2O4 was better than that of Co3O4 for activating PMS, which means doping Ni would help to improve the activity due to the redox reaction of Ni. In order to explore the adaptability, NiCo2O4/PMS system was used to degrade different pollutants, such as acid red 18 (AR18), alizarin red S (ARS), tetracycline (TC), and p-nitrophenol (PNP). The results were shown in Fig. S7 (Supporting information). Most dyes and tetracycline had high removal efficiency, however, p-nitrophenol was difficult to degrade.

|
Download:
|
| Fig. 3. (a) ESR spectrum of DMPO spin-trapping adducts, and (b) comparison of different systems. Experimental conditions: [RhB]0=100 mg/L, NiCo2O4 and Co3O4 doses=133.33 mg/L, T = 25 ℃, pH0 4.81. | |
{kind=link}
The intermediates of RhB were identified by LC–MS in Fig. S8 (Supporting information). The products with m/z values of 443, 432, 415, 354, 340, 274, 111, and 74 are obtained. The above results could speculate the pathway of rhodamine B degradation in NiCo2O4/PMS system in Fig. S9 (Supporting information).
Based on the above experiments and characterization, we can thus propose a reaction mechanism. As depicted in Fig. 4, the mechanism can be divided into two parts: A noninterference process and an interference process. When there is no interference, the redox reaction between Co2+/Ni2+ and Co3+/Ni3+ was generated on the surface of NiCo2O4 [39, 40], and PMS was activated to produce SO4·−. SO4·− will react with water, generating OH− and ·OH. Thus, SO4·− and ·OH coexisted in the reaction system. RhB was decomposed into CO2, H2O, and other byproducts. From the radical capture experiment and ESR, in our study, SO4·− and ·OH were the main active substances. When the system was interfered with inorganic anions, the process proceeded in two directions. HCO3−, NO3−, Cl− and H2PO4− would compete with RhB to obstruct the reaction. However, when the concentrations of Cl− and H2PO4− increased to 4000 ppm, reactive substances were generated, accelerating the reaction because of the formation of highly oxidizing species. The most important thing is that the active sites on the inner and outer surfaces can be utilized simultaneously, owing to the hole on the surface, which means that the mass transfer resistance of RhB, PMS, and product can be reduced. Therefore, the above two parts will occur simultaneously on the inner and outer surfaces, which can double the reaction area compared with other closed hollow materials.

|
Download:
|
| Fig. 4. Mechanism of NiCo2O4/PMS system. | |
{kind=link}
In conclusion, a recyclable open-type NiCo2O4 hollow-sphere was synthesized through a hydrothermal method with the assistance of a vesicle formed by NH3·H2O gas bubbles. The process of activating PMS to degrade RhB can occur simultaneously on the inner and outer surfaces, owing to the hole on the surface of our catalyst, which can reduce the mass transfer resistance of RhB, PMS, and products between the outer and the inner surface. Because of the double reaction area, during the degradation reaction, the removal rate can reach 100% and the concentration of pollutants is very high. Further research found that SO4·− and ·OH coexist in the reaction system. Finally, due to the complexity of polluted water, the interference of coexisting anions was studied. The results showed that low concentrations of HCO3−, NO3−, Cl− and H2PO4− will obstruct the reaction because of the generation of substances with low oxidation potential. When the concentration of Cl− and H2PO4− increased to 4000 ppm, reactive substances were generated to accelerate the reaction, due to the formation of highly oxidizing species. From this, we developed a potential catalyst for the remediation of organic pollutants.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Hebei Natural Science Foundation (No. B2020208064), the Double Tops Joint Fund of the Yunnan Science and Technology Bureau and Yunnan University (No. 2019FY003025), and Shijiazhuang Science and Technology Department (No. 191240263A).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.063.
| [1] |
K.A. Lin, J.T. Lin, Chemosphere 182 (2017) 54-64. DOI:10.1016/j.chemosphere.2017.04.152 |
| [2] |
M.H. Cui, L. Gao, H.S. Lee, A.J. Wang, Bioresour. Technol. 297 (2020) 122420. DOI:10.1016/j.biortech.2019.122420 |
| [3] |
P. Liu, L. Xing, H. Lin, et al., Sci. Bull. 62 (2017) 931-937. DOI:10.1016/j.scib.2017.05.031 |
| [4] |
J. Wu, Q. Li, W. Li, et al., J. Cleaner Prod. 251 (2020) 119694. DOI:10.1016/j.jclepro.2019.119694 |
| [5] |
L. Zeng, L. Xiao, X. Shi, et al., J. Colloid Interface Sci. 534 (2019) 586-594. DOI:10.1016/j.jcis.2018.09.074 |
| [6] |
M. Aleksić, H. Kušić, N. Koprivanac, D. Leszczynska, A.L. Božić, Desalination 257 (2010) 22-29. DOI:10.1016/j.desal.2010.03.016 |
| [7] |
X. Wang, Y. Pan, Z. Zhu, J. Wu, Chemosphere 117 (2014) 638-643. DOI:10.1016/j.chemosphere.2014.09.055 |
| [8] |
S. Rodríguez, D. Lorenzo, A. Santos, A. Romero, J. Environ. Manage. 255 (2020) 109926. DOI:10.1016/j.jenvman.2019.109926 |
| [9] |
B. Kakavandi, A. Takdastan, N. Jaafarzadeh, et al., J. Photochem. Photobiol. A 314 (2016) 178-188. DOI:10.1016/j.jphotochem.2015.08.008 |
| [10] |
J. Seo, H.J. Lee, H. Lee, et al., Chem. Eng. J. 273 (2015) 502-508. DOI:10.1016/j.cej.2015.03.114 |
| [11] |
M. Cheng, C. Lai, Y. Liu, et al., Coord. Chem. Rev. 368 (2018) 80-92. DOI:10.1016/j.ccr.2018.04.012 |
| [12] |
Y. Zhang, M. Zhou, J. Hazard. Mater. 362 (2019) 436-450. |
| [13] |
A.D. Bokare, W. Choi, J. Hazard. Mater. 275 (2014) 121-135. DOI:10.1016/j.jhazmat.2014.04.054 |
| [14] |
S. Hu, Y. Yu, Y. Guan, et al., Chin. Chem. Lett. 31 (2020) 2839-2842. DOI:10.1016/j.cclet.2020.08.021 |
| [15] |
H. Zhang, J. He, C. Zhai, M. Zhu, Chin. Chem. Lett. 30 (2019) 2338-2342. DOI:10.1016/j.cclet.2019.07.021 |
| [16] |
S. Wacławek, H.V. Lutze, K. Grübel, et al., Chem. Eng. J. 330 (2017) 44-62. DOI:10.1016/j.cej.2017.07.132 |
| [17] |
P. Hu, M. Long, Appl. Catal. B: Environ. 181 (2016) 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| [18] |
P. Shao, X. Duan, J. Xu, et al., J. Hazard. Mater. 322 (2017) 532-539. DOI:10.1016/j.jhazmat.2016.10.020 |
| [19] |
M. Zhang, J. He, Y. Chen, et al., Chin. Chem. Lett. 31 (2020) 2721-2724. DOI:10.1016/j.cclet.2020.05.001 |
| [20] |
Y. Pang, Y. Ruan, Y. Feng, et al., Chemosphere 228 (2019) 412-417. DOI:10.1016/j.chemosphere.2019.04.164 |
| [21] |
X. Lin, Y. Ma, J. Wan, Y. Wang, Y. Li, Appl. Catal. A 589 (2020) 117307. DOI:10.1016/j.apcata.2019.117307 |
| [22] |
Y. Zhao, G. Wang, L. Li, X. Dong, X. Zhang, Chemosphere 244 (2020) 125526. DOI:10.1016/j.chemosphere.2019.125526 |
| [23] |
Y. Li, L. Pan, Y. Zhu, et al., Water Res. 163 (2019) 114912. DOI:10.1016/j.watres.2019.114912 |
| [24] |
Z. Yang, Y. Li, X. Zhang, et al., Chem. Eng. J. 384 (2020) 123319. DOI:10.1016/j.cej.2019.123319 |
| [25] |
J. Sharma, I.M. Mishra, D.D. Dionysiou, V. Kumar, Chem. Eng. J. 276 (2015) 193-204. DOI:10.1016/j.cej.2015.04.021 |
| [26] |
X. Zhang, J. Yao, Z. Zhao, J. Liu, Chem. Eng. J. 364 (2019) 1-10. DOI:10.1145/3290605.3300242 |
| [27] |
S. Su, W. Guo, C. Yi, Y. Leng, Z. Ma, Ultrason. Sonochem. 19 (2012) 469-474. |
| [28] |
C. Cai, H. Zhang, X. Zhong, L. Hou, J. Hazard. Mater. 283 (2015) 70-79. |
| [29] |
H. Liu, P. Sun, M. Feng, et al., Appl. Catal. B: Environ. 187 (2016) 1-10. DOI:10.1080/13607863.2016.1269148 |
| [30] |
Y. Wang, S. Hui, S. Zhan, et al., Chem. Eng. J. 381 (2020) 122563. |
| [31] |
J. Zhou, W. Liu, W. Cai, Sci. Total Environ. 696 (2019) 133962. |
| [32] |
J. Li, M. Xu, G. Yao, B. Lai, Chem. Eng. J. 348 (2018) 1012-1024. |
| [33] |
Y. Ren, L. Lin, J. Ma, et al., Appl. Catal. B: Environ. 165 (2015) 572-578. |
| [34] |
W.D. Oh, V.W.C. Chang, Z.T. Hu, R. Goei, T.T. Lim, Chem. Eng. J. 323 (2017) 260-269. |
| [35] |
Z. Huang, Y. Yao, J. Lu, et al., J. Hazard. Mater. 301 (2016) 214-221. |
| [36] |
Y. Yao, Y. Cai, G. Wu, et al., J. Hazard. Mater. 296 (2015) 128-137. |
| [37] |
H. Zhang, C. Lu, H. Hou, Y. Ma, S. Yuan, Chem. Eng. J. 370 (2019) 400-408. |
| [38] |
T. Sharifi, C. Larsen, J. Wang, et al., Adv. Energy Mater. 6 (2016) 1600738. DOI:10.1002/aenm.201600738 |
| [39] |
W. Zhang, Y. Su, X. Zhang, Y. Yang, X. Guo, RSC Adv. 6 (2016) 64626-64633. DOI:10.1039/C6RA12706A |
| [40] |
X. Tian, C. Tian, Y. Nie, et al., Chem. Eng. J. 331 (2018) 144-151. |
| [41] |
C. Jiang, Y. Ji, Y. Shi, J. Chen, T. Cai, Water Res. 106 (2016) 507-517. |