 2021, Vol. 32
2021, Vol. 32 



b Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University & Collaborative Innovation Centre of Chemical Science and Engineering, Tianjin 300072, China;
c Centre for Programmable Materials, School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore;
d Hanshan Normal University, Chaozhou 521041, China;
e ZheJiang Fenghong New Material Co., Ltd., Huzhou 313300, China
Low-temperature electrochemical methanol oxidation reaction (MOR) plays a crucial role in direct liquid fuel cells (DLFCs) and electrochemical synthesis [1]. To date, metal has been demonstrated to show excellent electrochemical methanol oxidation activity owing to its favourable electronic structure and high electrical conductivity [2-8]. Especially metallic palladium (Pd) is commonly used in MOR. However, pure Pd nanoparticles (NPs) catalysts usually suffer from quick poisoning by reaction intermediates, which block the catalytically active sites and cause dissolution and aggregation of Pd active species during the catalytic process, leading to decreased activity and long-term instability [9, 10]. To solve this problem, the rational design and synthesis of heterojunction has been considered as an effective strategy to enhance both the dispersity and stability of the catalytically active species, and thus improve the catalytic performance [11-17]. The interface of heterojunction has been widely used in many areas, including oxygen reduction reaction, electrocatalytic CO2 reduction in renewable energy storage and conversion systems [18-21].
Traditional metal-support heterojunctions, i.e., metal NPs (e.g., Pd, Pt, Au, Cu, ZnPd) supported on metal oxides, particularly reducible oxides such as TiO2, CeO2, and FeOx, CeOx, Co3O4, MoC, have been demonstrated to be an effective strategy to improve the catalytic performance with high activity and or selectivity owing to their strong metal-support interaction (SMSI) effect [22-33]. However, catalytic reactions are often suppressed when the metal active sites are blocked in the classic prototype in metal-support interactions, which would severely district or limit its large-scale application and lifetime [34]. Different from traditional heterogeneous catalyst, an inverse heterogeneous catalyst, in which the metal surface is decorated by metal oxide nano islands under reducing conditions has recently attracted increasing attention in the catalytic hydrogenation or dehydrogenation reactions, oxidative reaction and electrochemical reaction [24, 35, 36]. It would cause immense environmental and public interest to develop inverse heterogeneous metal NPs partially covered by metal oxides since the high atomic utilization and recyclability [36, 37]. Metal oxides could not only act as a carrier to prevent the sintering of metal NPs, but also have a great influence on the catalytic activity of the catalyst. The cause of this phenomenon is motivated by reducing of the high surface energy of the metal catalysts. The surface oxides could modulate the local electronic structure of the noble metal inside and it can be used as Lewis acids to activate the reactants and enhance activity of the catalyst. However, the surface coverage is not as high as possible. Extensive surface coverage of metal oxide on noble metal has a fatal effect on the catalytic activity owing to blockage of the active center. In an inverse heterogeneous catalyst, the metal cations at the edges of metal oxide are coordinatively unsaturated, which could be observed by scanning tunneling microscopy (STM) and high angle annular dark fieldscanning transmission electron microscopy (HAADF-STEM), further creates high density oxide edge active sites [37, 38]. Coordinative unsaturated transition metal (CUTM) sites confined in the interface between a transition metal oxide and a noble metal are highly active centers which are able to promote catalytic reactions in a wide range of heterogeneous catalytic reactions [39, 40]. Thus, the synergetic effect could come from both metal oxide edge and noble metal surface sites, thus which are able to generate interfacial dual active sites [35]. Generally, the reaction active species is activated oxygen which could be readily obtained from interfacial dissociated oxygen gas, which further participates in the catalytic reaction [41, 42]. Bao and co-workers discovered that the exposed ferrous centers at the interface FeO/Pt catalysts are important active sites contributing to the enhancement of catalytic oxidation manifested by STM characterization and DFT calculation [27]. Li et al. and co-workers reported the dual active sites of the FeO/Pt(111) interface in a gas reaction [36]. However, until now, rare research efforts have been devoted to the study of the interaction and the nature of the active species on inverse heterogeneous catalyst from the aspect of atomic, electronic, geometric and electrochemical reaction pathways viewpoint.
Herein, an inverse Fe2O3/Pd hybrid structure was taken as an example for electrochemical MOR by density functional theory (DFT). We modify the electronic (charge transfer between the metal sites and metal oxide) and/or geometric (decoration or coverage of metal oxide) parameters, and also the electrochemical reaction pathways, e.g., interfacial hydroxyl-assisted catalytic reaction pathway. The Pd-Fe2O3 structural mismatch, the nature of the interface bonding, charge transfer and work function were calculated. The Fe2O3/Pd is found more energetically favorable as compared to bare Pd for selective MOR to formaldehyde by the adsorbed hydroxyl (OH*) species, which gives a clear picture of how the interface promotes catalytic reaction in the Fe2O3/Pd system. This work could guide the experimentalist to improve the utilization rate of precious metal in the catalyst and maximize the active sites of the catalysts.
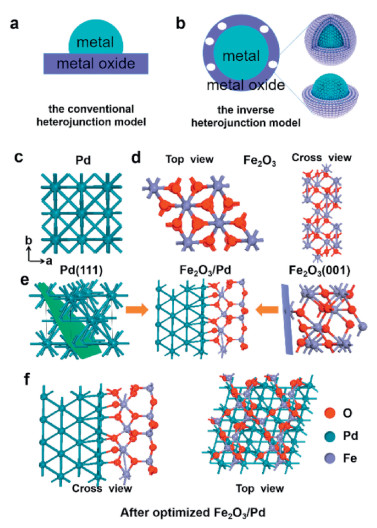
Traditionally, these catalysts were synthesized by anchoring active metal nanoparticles on some high specific surface area carriers to increase the dispersion of catalytic active sites and stabilize metal against leaching in Fig. 1a. A reverse metal–support interactions is defined as the encapsulation of NPs by partially reduced oxide supports. Ideally, the oxide coating on the metal surface should be very thin and permeable to small molecules, while still completely wrapping metal nanoparticles to prevent the dissolution, disintegration and aggregation of active centers in the catalytic process shown in Fig. 1b. Inverse heterojunction is unique and rarely studied. Figs. 1c and d give the atomic model of bulk Pd and Fe2O3 structure with top and cross-sectional views. The lattice constants for bulk Pd and Fe2O3 are a = b = c=3.895 and a = b =5.928 Å, c =35.69 Å with an angle of 120.0°, respectively. The Pd(111) and Fe2O3(001) were selected to construct a heterojunction model owing to a small planar mismatch as displayed in Fig. 1e and magnified by XRD and HR-STEM (lattice parameter of 0.217 nm) characterizations by Zhu et al. [35]. To identify the most stable heterojunction model, Fe2O3 is shifted along the x-direction from 0 to 1.2 Å, giving a lowest formation energy at 0.4 Å. Subsequently, a whole structure relaxation was performed according to the optimized structure with the interface configuration of Fe2O3/Pd as shown in Fig. 1f. At the Fe2O3/Pd interface, each oxygen atom is bonded to one Pd atom with Pd-O bond length of 1.89 Å and 1.92 Å and two Fe atoms with Fe-O bond length of 2.00 Å. Interfacial Fe-O-Pd angle is 136.71°. From top view, the exposed O and Fe atoms are located on the same plane, which is different from the bulk structure. Furthermore, both Fe-O3-Fe and O-Fe-O3 terminations have been shown stable, which coexist on exposed α-Fe2O3(0001) facet [43].

|
Download:
|
| Fig. 1. Schematic illustration of conventional heterojunction model (a) and inverse heterojunction with half-covered model (b). Top view of Pd (c) top and cross-sectional view of Fe2O3 (d) and schematic construction of the interface between Pd and Fe2O3 (e). Top and cross-sectional configurations of Fe2O3/Pd heterojunction (f). Blue, purple and red balls denote Pd, Fe and O atoms, respectively. | |
{kind=link}
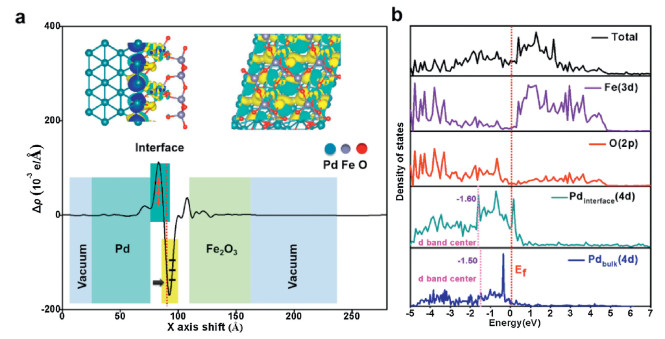
To explore charge distribution across the interface of Fe2O3/Pd, charge transfer between Pd and Fe2O3 was calculated by subtracting the electronic charge of a Fe2O3/Pd composite from that of pure Pd and Fe2O3 and the result is shown in Fig. 2a. The upper figure displays a three-dimensional atomic model after optimization. At the interface between Pd and Fe 2O3, the cyan region on Pd indicates charge depletion, while the yellow region on Fe2O3 shows charge accumulation. The charge transfer only occurs at the interface owing to the strong interaction between Pd and Fe2O3. The charge depletion and charge accumulation constitute the interfacial charge redistribution, resulting in the formation of interfacial electric dipole. The charge density difference result shows that electrons mainly transfer from Pd side to Fe2O3 side, which is consistent with the observation of X-ray photoelectron spectroscopy (XPS) illustrating a positively charged Pd (Pdδ+) and CO diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) (CO adsorption on positively charged Pd (CO-Pdδ+)) [44]. In Fig. S1 (Supporting information), when the thickness of Fe2O3 is increased double, it is found that the direction of charge transfer is opposite to that of thin Fe2O3, that is, from Fe2O3 to Pd, which rationalize that too much thickness Fe2O3 is detrimental to the MOR performance. The d band center is a vital criterial to affect the CH3OH intermediate species adsorption ability, further affects CH3OH oxidation reaction, thus it is essential to obtain this data by calculating the density of states (DOS). Fig. 2b shows the total density of states (TDOS) and partial density of states (PDOS) on Fe2O3/Pd. Three features are apparent: (Ⅰ) There is no gap in the entire slab as shown in the TDOS. (Ⅱ) Oxygen atoms mainly constitute the valance band maximum (VBM) and Fe atoms contribute to the conduction band minimum (CBM). (Ⅲ) The d band center (pink line, −1.6 eV) of Pd in Fe2O3/Pd is downshifted with respect to that of Pd particles (−1.5 eV). The interface without band gap will not trap the carriers and consequently avoids Fermi level pinning. According to the theory proposed by Nørskov, the d band center downshift implies the Pd-Fe-O active centers in Pd/Fe2O3 composite guarantee an appropriate adsorption for the intermediates in methanol selective oxidation, thereby significantly promoting the methanol selective oxidation to formaldehyde process. What is more, the bands near fermi energy have more orbitals which could enhance the interaction between adsorption intermediate and base Pd.

|
Download:
|
| Fig. 2. (a) Charge density difference (Δρ = ρ(Pd-Fe2O3) - ρ(Pd) - ρ(Fe2O3)) of Pd-Fe2O3. The yellow region represents charge accumulation, and the cyan region indicates charge depletion. The blue balls represent Pd, purple balls represent Fe and the red balls represent O atoms, respectively. (b) The total and partial density of states of Fe2O3/Pd and Pd particles. Red dashed line shows Fermi energy and pink dashed line represents d band center. | |
{kind=link}
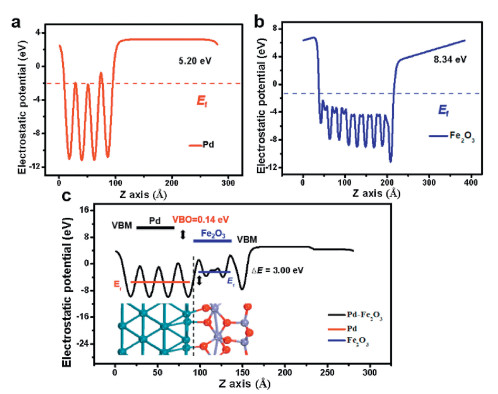
The quantitative calculation of the change in the dipole moments at these interfaces is meaningful. To investigate the built-in potential at the Fe2O3/Pd interface, the electrostatic potential along the z normal, namely the work-function, was computed as shown in Fig. 3. The calculated electrostatic potential of pure Pd and Fe2O3 is 5.20 and 8.34 eV, respectively. The average potential on the Fe2O3/Pd heterojunction of the Fe2O3 side is higher than that of the Pd side with an electrostatic potential difference of 3.00 eV, which is smaller than the difference (3.14 eV) between pure Pd and Fe2O3.

|
Download:
|
| Fig. 3. Electrostatic potential of pure (a) Pd, (b) Fe2O3 and (c) Fe2O3/Pd heterojunction along the z-axis, with the red and blue line representing the average potential of Pd and Fe2O3, respectively. | |
{kind=link}
Here, the valance band offset (VBO) is built-in potential which could drive the electrons transfer. VBO could be calculated by potential-line-up method according to the formula:
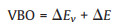
|
where ΔE is referred to the difference of the VBM obtained from two independent standard bulk band structure calculations at the same strained geometries as in the supercell calculation. ΔE results from the line-up of the macroscopic average of the self-consistent electrostatic potential across the interface. The deduced VBO was calculated to be approximately 0.14 eV. Thus, the driving force direction of electron flow is from Pd to Fe2O3 (Fig. 3c), rationalizing the charge transfer induced charge density difference from the qualitative and quantitative aspects as displayed in Fig. 2a.
The above-mentioned interface is coordinated saturated bonding in an ideal system, which is different from traditional heterogeneous Pd NPs decorated on Fe2O3 structure (Fig. S2 in Supporting information). While the other situation is the uncoordinated interfacial arrangement which may serve as the catalytic active site for MOR. Previous Extended X-ray Absorption Fine Structure (EXAFS) curve fitting combined with DFT simulations were carried out to provide more insight into the chemical bond between Pd and Fe2O3 which has the strong metal–support interaction via a reverse route (SMSIR). The result found that Pd-Fe bond formation would promote the reconstruction of the interface, the rearrangement of oxide lattice and the mobility of Pd atoms [38, 44]. In our work, DFT investigated the interfacial atomic arrangement and terminals of Fe2O3/Pd in detail. In Fig. S3 (Supporting information), three structures comprising of different and terminal atom were investigated: (1) With oxygen atoms as interfacial contacting atoms and oxygen atoms as terminal atom (Figs. S3a and b); (2) with oxygen atoms as interfacial contacting atoms and Fe as terminal atom (Figs. S3c and d); (3) with Pd-Fe-O as interfacial contacting atoms and the coordination-unsaturated Fe as terminal atom (Figs. S3e and f). The interfacial binding energy is calculated according to 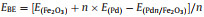

|
Download:
|
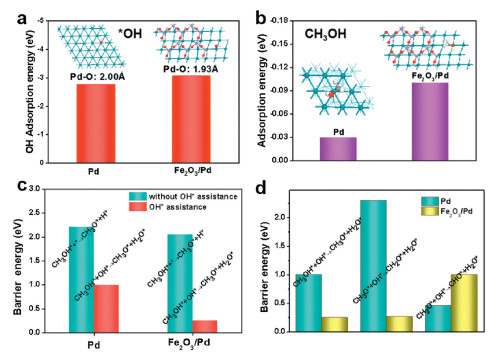
| Fig. 4. Comparison of (a) OH* adsorption energy and (b) CH3OH adsorption on pure Pd and Fe2O3/Pd. (c) Comparison of the activation barrier for the first dehydrogenation steps in methanol oxidation on Pd(111) and Fe2O3/Pd surfaces with or without hydroxyl OH* assistance. (d) Comparison of the activation barrier for the first three dehydrogenation steps in methanol oxidation reaction on Pd(111) and Fe2O3/Pd surfaces with hydroxyl OH* assistance. | |
{kind=link}
We conducted a thermodynamic study on the pathway of methanol catalytic reaction on pure Pd and Fe2O3/Pd surfaces with assistant of hydroxyl by DFT. The reaction path involves solution-mediated and metal-catalysed elementary steps. In the decomposition methanol process shown in Fig. S4 (Supporting information), there exist two possibilities to dehydrogenation: C-H bond scission (black line) and the O-H bond cleavage alternatively (red line). The calculated data of barrier energy for Pd and Fe2O3/Pd is shown in Table S2 (Supporting information) and Figs. S5 and S6 (Supporting information). The road of Pd would like to react along pathway 1, illustrating C-H bond cleavage is more feasible than O-H bond scission owing to lower barrier energy. While on the Fe2O3/Pd, reaction energy of CH3OH decomposition energetically favors O-H bond scission with a barrier energy of 2.05 eV, which is smaller than that C-H bond cleavage (2.61 eV) shown in Fig. S6. In all, free of hydroxyl OH*, the energy barrier of the catalytic reaction is relatively high, and the trend is upward, which is fatal to the MOR catalysis. While, in the alkaline electrochemical conditions, hydroxyl OH* in solution would assist heterogeneous catalyst dehydrogenation reactions of CH3OH, which is different from gas phase reaction [48, 49]. The reaction Gibbs free energy of MOR on Pd(111) and Fe2O3/Pd surfaces with an assistance of interfacial hydroxyl OH* are investigated following the reaction pathway and configurations shown in Fig. S7 (Supporting information). The result shows inverse heterogeneous Fe2O3/Pd has lower Gibbs free energy than pure Pd. As shown in Fig. 4c and Table S3 (detail configurations and barrier energy in Supporting information), the calculated dissociative barrier energy of methanol on the Pd surface is as high as 2.21 eV, and thus the O-H bond of CH3OH activation by Pd is unlikely. However, the presence of surface hydroxyl OH* intermediate can facilitate the O-H bond (in methanol) activation via the proton transfer in nearly the same way as it occurs in solution. This process significantly lowers the activation barrier to less than 1.00 eV, which is smaller than without OH assistant. The volcano map with configurations is shown in Fig. S8 (Supporting information). Moreover, the calculated energy barrier of the first dehydrogenation step (CH3OH* + OH* → CH3O* + H2O*) on Fe2O3/Pd is as low as 0.25 eV. As shown in Fig. 4d, the energy barrier of the second dehydrogenation step (CH2OH* + OH* → CH3O* + H2O*) on Fe2O3/Pd is 0.27 eV, while second dehydrogenation step on Pd is 2.30 eV. However, the energy barrier of the third dehydrogenation step (CH2O* + OH*→ CHO* + H2O*) shows the opposite result, which is larger on Fe2O3/Pd compared to that on Pd. These results suggest that formed formaldehyde on Fe2O3/Pd could not be easily further dehydrogenated, leading to a highly selective MOR to formaldehyde on the Fe2O3/Pd surface. The formation of the Fe2O3/Pd interface would induce electron transfer from Pd to Fe2O3, which could downshift the d band centre of Pd, thus increases the surface reactivity and enhances the adsorption ability of hydroxyl OH*, greatly facilitating the process of selective methanol oxidation to formaldehyde. We calculated the reaction free energy using the computational hydrogen electrode (CHE) method. The MOR on Fe2O3/Pd is more feasible than Pd from Gibbs free energy points. The MOR pathway on Fe2O3/Pd undergoes i.e., CH3OH* + OH* → CH3O* + OH* → CH2O* + OH* → CHO* + OH*, with the lower Gibbs free energy. Intermediates including CH3OH*, CH3O*, CH2O*, CHO*, are all adsorbed on Pd side of Fe2O3/Pd; Fig. S9 (Supporting information) show the optimize structures of all active species involved in MOR on Pd(111). Because the electronegativity of Fe (1.83) is much lower than Pd (2.20) [38], the alloying of Fe can modify the Pd electronic properties as the electrons will transfer from Pd to Fe and contribute to thed band hybridization of Pd and Fe. The downward shift of the d band center relative to the Fermi level would enhance the adsorption of OH* species on the Pd surface and greatly facilitate the process of MOR. It is reasonable that the Fe2O3 will have a positive effect on the electrocatalytic Pd NPs. However, excess amount of Fe2O3 amount can largely decrease the electrocatalytic activity due to its poor conductivity. A clear picture of how the interface promotes MOR catalytic reaction in the Fe2O3/Pd system shown in Fig. S10 (Supporting information).
In summary, this work demonstrates that the inverse metal-support interface plays an important role in low-temperature electrochemical MOR based on the investigation of the Fe2O3/Pd interface by first-principles calculations. It is found that (Ⅰ) inverse Fe2O3 partially covering Pd system could exhibit better MOR performance than Pd and could highly electrochemically transform methanol into formaldehyde in reaction route CH3OH* → CH3O* → CH2O* → CHO* via two electrons transferring based on the calculated adsorption energy and transition state barrier energy. (Ⅱ) The charge could transfer from Pd to Fe2O3 driven by the built-in potential at the interface of Pd and Fe2O3. (Ⅲ) The Fe2O3/Pd heterostructure can beneficially lower the d band centre of Pd in Fe2O3/Pd, which thus promotes adsorption of CH3OH on the Pd sites. (Ⅳ) Interfacial hydroxyl between Fe2O3 and Pd plays an important and valuable role in MOR compared with free of hydroxyl-assistance, which could lower the barrier energy distinctly. All the above contributions effectively reduce the activation barrier energy of the electrochemical MOR on Fe2O3/Pd. This atomic and electronic calculation investigation provides a general and universal heterogeneous insight, which could be also applied to other metals (e.g., Pt, Au, Rh) and metal oxides (e.g., FeOx, NiOx, CoOx).
Declaration of competing interestThe authors claim that no conflict of interest exists in the submission of this manuscript, and manuscript is approved by all authors for publication.
AcknowledgmentsThe work was financially supported by the National Natural Science Foundation of China (Nos. 21805191, 21866032), the Guangdong Basic and Applied Basic Research Foundation (No. 2020A151501982) and the China Postdoctoral Science Foundation (No. 2020M672811). The work is completed on the "Explorer 100" cluster system of Tsinghua National Laboratory for Information Science and Technology.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.057.
| [1] |
A.P.V. Soares, M.F. Portela, A. Kiennemann, Catal. Rev. 47 (2005) 125-174. DOI:10.1081/CR-200049088 |
| [2] |
F. Nosheen, N. Wasfi, S. Aslam, et al., Nanoscale 12 (2020) 4219-4237. DOI:10.1039/C9NR09557H |
| [3] |
Y. Zhang, J. Zhang, Z. Chen, et al., Sci. China Mater. 61 (2018) 697-706. DOI:10.1007/s40843-017-9157-9 |
| [4] |
Z. Tao, W. Chen, J. Yang, et al., Sci. China Mater. 62 (2019) 273-282. DOI:10.1007/s40843-018-9366-x |
| [5] |
A. Shan, S. Huang, H. Zhao, et al., Nano Res. 13 (2020) 3088-3097. DOI:10.1007/s12274-020-2978-3 |
| [6] |
Y. Sun, Y. Zhou, Y. Liu, et al., Nano Res. 13 (2020) 2683-2690. DOI:10.1007/s12274-020-2910-x |
| [7] |
W. Lei, M. Li, L. He, et al., Nano Res. 13 (2020) 638-645. DOI:10.1007/s12274-020-2666-3 |
| [8] |
J.C. Guo, Y.F. Lin, N. Tian, et al., Acta Phys.-Chim. Sin. 35 (2019) 749-754. DOI:10.3866/PKU.WHXB201810051 |
| [9] |
L.C. Kao, A.C. Hutson, A. Sen, J. Am. Chem. Soc. 113 (1991) 700-701. DOI:10.1021/ja00002a063 |
| [10] |
J.C. Colmenares, P. Lisowski, D. Łomot, O. Chernyayeva, D. Lisovytskiy, ChemSusChem. 8 (2015) 1676-1685. DOI:10.1002/cssc.201403125 |
| [11] |
Q.Q. Shi, G.C. Ping, X.J. Wang, et al., J. Mater. Chem. A 7 (2019) 2253-2260. DOI:10.1039/C8TA09439J |
| [12] |
H.Y. Kim, G. Henkelman, J. Phys. Chem. Lett. 4 (2013) 216-221. DOI:10.1021/jz301778b |
| [13] |
T. Kwon, M. Jun, J. Joo, K. Lee, J. Mater. Chem. A 7 (2019) 5090-5110. DOI:10.1039/C8TA09494B |
| [14] |
K. Shin, L. Zhang, H. An, et al., Nanoscale 9 (2017) 5244-5253. DOI:10.1039/C7NR01382E |
| [15] |
Q.C. Feng, S. Zhao, D.S. He, et al., J. Am. Chem. Soc. 140 (2018) 2773-2776. DOI:10.1021/jacs.7b13612 |
| [16] |
Z.C. Zhang, G.G. Liu, X.Y. Cui, et al., Adv. Mater. 30 (2018) 1801741. DOI:10.1002/adma.201801741 |
| [17] |
Z.C. Zhang, B. Xu, X. Wang, Chem. Soc. Rev. 43 (2014) 7870-7886. DOI:10.1039/C3CS60389J |
| [18] |
S. Wan, J. Wu, D. Wang, et al., Chin. Chem. Lett. 32 (2021) 816-821. DOI:10.1016/j.cclet.2020.04.040 |
| [19] |
C.H. Yang, F. Nosheen, Z.C. Zhang, Rare. Met. 40 (2021) 1412-1430. DOI:10.1007/s12598-020-01600-4 |
| [20] |
C.L. Lv, C.H. Yang, L.Y. Liu, Z.C. Zhang, Rare. Met. 32 (2021) 1437-1442. |
| [21] |
C. Yang, S. Li, Z. Zhang, et al., Small 16 (2020) e2001847. DOI:10.1002/smll.202001847 |
| [22] |
H. Yang, B. Xu, Q.T. Zhang, et al., Appl. Catal. B 286 (2021) 119845. DOI:10.1016/j.apcatb.2020.119845 |
| [23] |
R.J. Davies, M. Bowker, P.R. Davies, D.J. Morgan, Nanoscale 5 (2013) 9018-9022. DOI:10.1039/c3nr03047d |
| [24] |
J.H. Dong, Q. Fu, Z. Jiang, B.B. Mei, X.H. Bao, J. Am. Chem. Soc. 140 (2018) 13808-13816. DOI:10.1021/jacs.8b08246 |
| [25] |
X.J. Xu, Q. Fu, L. Gan, J. Zhu, X.H. Bao, J. Phys. Chem. B 122 (2018) 984-990. DOI:10.1021/acs.jpcb.7b07644 |
| [26] |
X. Liu, M.H. Liu, Y.C. Luo, et al., J. Am. Chem. Soc. 134 (2012) 10251-10258. DOI:10.1021/ja3033235 |
| [27] |
F.L. Liao, T.W.B. Lo, J. Qu, et al., Catal. Sci. Technol. 5 (2015) 3491-3495. DOI:10.1039/C5CY00572H |
| [28] |
Q. Fu, W.X. Li, Y. Yao, et al., Science 328 (2010) 1141-1144. DOI:10.1126/science.1188267 |
| [29] |
X.J. Cui, W. Li, P. Ryabchuk, K. Junge, M. Beller, Nat. Catal. 1 (2018) 385-397. DOI:10.1038/s41929-018-0090-9 |
| [30] |
A. Chen, X.J. Yu, Y. Zhou, et al., Nat. Catal. 2 (2019) 334-341. DOI:10.1038/s41929-019-0226-6 |
| [31] |
N.J. O'Connor, A.S.M. Jonayat, M.J. Janik, T.P. Senftle, Nat. Catal. 1 (2018) 531-539. DOI:10.1038/s41929-018-0094-5 |
| [32] |
L.L. Lin, W. Zhou, R. Gao, et al., Nature 544 (2017) 80-83. DOI:10.1038/nature21672 |
| [33] |
Z. Li, Y.R. Cui, Z.W. Wu, et al., Nat. Catal. 1 (2018) 349-355. DOI:10.1038/s41929-018-0067-8 |
| [34] |
K. Shen, J.P. Lin, Q. Xia, et al., Rare. Met. 38 (2019) 107-114. DOI:10.1007/s12598-018-1081-3 |
| [35] |
Y.N. Sun, L. Giordano, J. Goniakowski, et al., Angew. Chem. Int. Ed. 49 (2010) 4418-4421. DOI:10.1002/anie.201000437 |
| [36] |
P.W. Wu, S. Tan, J. Moon, Z.H. Yan, H.Y. Zhu, Nat. Commun. 11 (2020) 3042. DOI:10.1038/s41467-020-16674-y |
| [37] |
G.F. Zhao, F. Yang, Z.J. Chen, et al., Nat. Commun. 8 (2017) 14039. DOI:10.1038/ncomms14039 |
| [38] |
Z.J. Chen, Y. Mao, J.F. Chen, et al., ACS Catal. 7 (2017) 4281-4290. DOI:10.1021/acscatal.7b00541 |
| [39] |
F.L. Liao, T.W.B. Lo, D. Sexton, et al., Catal. Sci. Technol. 5 (2015) 887-896. DOI:10.1039/C4CY01159G |
| [40] |
L. Cao, W. Liu, Q.Q. Luo, et al., Nature 565 (2019) 631-635. DOI:10.1038/s41586-018-0869-5 |
| [41] |
T.W. Deelen, C.H. Mejía, K.P. Jong, Nat. Catal. 2 (2019) 955-970. |
| [42] |
G.X. Chen, Y. Zhao, G. Fu, et al., Science 344 (2014) 495-499. DOI:10.1126/science.1252553 |
| [43] |
P.X. Liu, Y. Zhao, R.X. Qin, et al., Sci. Bull. 63 (2018) 675-682. DOI:10.1016/j.scib.2018.03.002 |
| [44] |
J.J. Tang, B. Liu, J. Phys. Chem. C 120 (2016) 6642-6650. DOI:10.1021/acs.jpcc.6b00374 |
| [45] |
C.T. Wu, K.M. Yu, F. Liao, et al., Nat. Commun. 3 (2012) 1050. DOI:10.1038/ncomms2053 |
| [46] |
C. Liu, Z.L. Chen, D.W. Rao, et al., Sci. China Mater. 63 (2020) 2095-8226. |
| [47] |
B.N. Zope, D.D. Hibbitts, M. Neurock, R.J. Davis, Science 330 (2010) 74-78. |
| [48] |
G.M. Mullen, L. Zhang, E.J. Evans, et al., Phys. Chem. Chem. Phys. 147 (2015) 4730-4738. |
| [49] |
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. |