 2021, Vol. 32
2021, Vol. 32 

b National & Local United Engineering Lab for Personalized Anti-tumor Drugs, The State Key Laboratory of Chemical Oncogenomics, Key Laboratory of Chemical Biology, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China;
c Institute of Biomedical Health Technology and Engineering, Shenzhen Bay Laboratory, Shenzhen 518055, China;
d National & Local United Engineering Lab for Personalized Anti-tumor Drugs, Shenzhen Kivita Innovative Drug Discovery Institute, Shenzhen 518110, China;
e School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China
Epigenetic control of gene expression plays a significant role in a variety of diseases, such as neurological disease, blood disorders, viral infection, diabetes, fibrosis and especially cancers. Over the past few years, researches on the mechanisms of epigenetics have explored, providing new insights into the role of epigenetic control in pathobiology and leading to the discovery of lots of specific drug targets, as well as facilitating the development of epigenetic medicinal chemistry [1-3]. DNA methyltransferase (DNMT) and histone deacetylase (HDAC) have long been two of the most attractive epigenetic targets that possess practical therapeutic potential since the very beginning of the epigenetic drug discovery [4].
In human cancers, aberrant expressed DNMT1 and DNMT3A/3B respectively catalyze the maintenance methylation and de novo methylation of cytosine in DNA CpG islands, leading to tumor suppressor genes (TSG) silencing [5]. Similarly, HDAC is responsible for the hypoacetylation of histones in nucleosomes, inducing chromatin remodeling and thereby hindering gene transcription. Besides, HDAC also affect a variety of cell functions by regulating non-histone substrates, including key tumor suppressor proteins and oncogenes [6]. Lots of DNMT inhibitors (DNMTi) and HDAC inhibitors (HDACi) have been developed [7-18] as antitumor agents being able to modulate enzymatic activity and reverse cancer cells' malignant phenotype.
Both DNMT and HDAC can induce TSGs silencing. Moreover, the two enzymes are closely related by interact with each other directly or via cofactors [19]. The interplay/crosstalk of DNMT and HDAC gives rise to inherent self-reinforcing nature of silencing mechanisms, and hence it could cause drug resistance by bypass compensatory mechanism when DNMTi or HDACi are used as single agents [20]. On the other side, precious studies indicated that combination therapy consisting of DNMTi and HDACi displayed significant synergistic effect and greatly improved anticancer activity [21-23]. In the past few years, we have constructed hydroxamic acid derivatives of NSC-319725 and carbazole to develop dual DNMT and HDAC inhibitors. The representative compound 15a [24] and C02S [25] indicated that targeting DNMT and HDAC simultaneously could be an effective strategy to develop antitumor agents. However, the DNMT inhibitory potency of 15a (70% inhibition rate against DNMT1 at 100 μmol/L) and the HDAC inhibitory activity of C02S [HDAC1 half maximal inhibitory concentration (IC50) = 4.16 μmol/L] need to be further optimized.
To advance more potent DNMT and HDAC dual inhibitors, in this report we designed and synthesized a novel series of hydroxamic acid derivatives of nucleoside bases (e.g., cytosine, uracil and adenine) utilizing fragment-based rational drug design strategy. The unique pharmacologic properties of representative compound 204 and its potential anticancer potency through epigenetic reprogramming were documented.
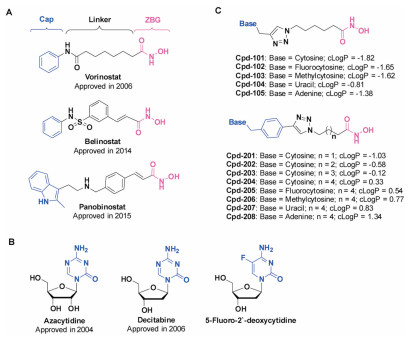
Generally, HDACi could be divided into different classes (e.g., hydroxamates, cyclic peptides, benzamides and fatty acids) according to their chemical structures. HDACi possessing hydroxamic acid moiety have in common a well-admitted pharmacophore model and share similar characteristics consisting of three groups: a cap group, which occludes the entrance of the active site pocket; a zinc-binding group (ZBG), which chelates the zinc ion in the active site and is required for catalytic function; and a linker, which connects the cap group and ZBG [26-28]. Now three hadroxamates (i.e., vorinostat, belinostat and panobinostat) have been approved by the US FDA (Fig. 1A). On the other side, DNMTi usually divided into nucleoside analogues (e.g., azacytidine and decitabine) (Fig. 1B) and non-nucleoside analogues (e.g., RG108, SGI1027 and DC-517) according to their chemical structures and functional mechanisms. Nucleoside analogues exhibited potent DNMT inhibitory potency and significant antitumor activity, while they could also cause advance side effects by incorporating into DNA or RNA.

|
Download:
|
| Fig. 1. (A) Approved hydroxamates as HDAC inhibitors; (B) Reported nucleoside analogues as DNMT inhibitors; and (C) Proposed dual DNMT and HDAC inhibitors. cLogP was calculated using DataWarrior prediction. | |
Given that cytosine is a component part of the substrate of DNMT, and that the approved DNMTi azacytidine and decitabine are derivatives of cytosine, herein we intended to develop dual DNMT and HDAC inhibitors by incorporating the hydroxamic acid group to cytosine through a proper linker unit (Fig. 1C). We proposed that the cytosine groups of our designed compounds could occupy the substrate binding domain of DNMT, therefore the compounds might possess DNMT inhibitory activity and cause less side effects than nucleoside analogues for they would not incorporate into DNA/RNA. Besides, the compounds might display HDAC inhibitory activity as they contain the three pharmacophore characteristics of HDAC inhibitors: a cap group, a zinc-binding group and a linker. Additionally, uracil and adenine hydroxamic acid derivatives were also constructed as compared compounds.
We initially constructed a triazole cycle to connect nucleoside bases and hydroxamic acid group for the sake of convenient synthesis (Fig. 1C). Unfortunately the obtained compounds (e.g., Cpd-101) showed too strong water solubility with unfavorable cLogP values to be used in medical or biological applications. To circumvent this issue, we then optimized the linker by inserting a benzene group, which could also be a part of the cap to compose HDACi. The resulted compounds 201–208 showed more favorable cLogP values than compounds 101–105 (Fig. 1C). We then synthesized the designed compounds following procedures showed in Scheme 1. The detailed synthesis and characterization of final products 101–105 and 201–208 were documented in Supporting information.

|
Download:
|
| Scheme 1. Synthesis of compounds (A) 101–105 and (B) 201–208. Reagents and conditions: (a) K2CO3, DMF; or Bu4NOH, DCM; (b) NaN3, DMF, 80℃; (c) sodium ascorbate, CuSO4 ·5H2O, t-BuOH/H2O, 60℃; (d) NH2OH, NaOCH3, MeOH. (e) trimethylsilylacetylene, Pd(PPh3)2Cl2, CuI, Et3N, THF, 50℃, 18 h; (f) K2CO3, MeOH, 2 h; (g) LiAlH4, THF, 0℃ to r.t.; (h) MsCl, Et3N, DCM; (i) Cs2CO3, DMF. (C) Structures of reagents 1a–1e. | |
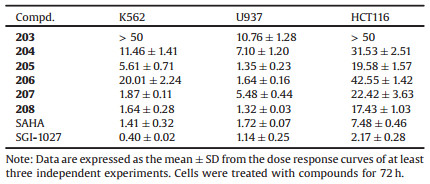
To get potential antitumor agents, we firstly evaluated the inhibitory activities of our compounds against tumor cells proliferation, using HDAC inhibitor SAHA and DNMT inhibitor SGI1027 as positive compounds. The inhibition rates of tumor cells treating with our compounds were primarily tested. The results indicated that most of our compounds (especially 101–105, 201 and 202) showed pretty weak proliferation inhibitory activity (inhibition rates less than 50%) in solid tumor cells, such as A549, HCT116, HeLa and MDA-MB-231, but showed better inhibitory activity in leukemia cells, such as K562 and U937 (Fig. S1 in Supporting information). The weak activity of our compounds 101–105, 201 and 202 might be caused by the too strong hydrophilic property to enter tumor cells. We further evaluated the IC50 values of compounds 203–208, which showed > 50% inhibition rate in tumor cells K562, U937 and HCT116. As shown in Table 1, most compounds showed more potent inhibitory activity against U937 than K562 and HCT116. Compounds 204–208 could significantly inhibited U937 cells proliferation with IC50 values less than 10 μmol/L.
|
|
Table 1 Cytotoxicity to human cancer cells (IC50, μmol/L). |
As compounds 203–208 showed good antitumor proliferation activity, we then evaluated the DNMT and HDAC inhibitory potency of compounds 201–208. As shown in Table 2, compounds 201–204 exhibited potent inhibitory activity against DNMT1 (inhibition rates approximate to 90%) and barely inhibited DNMT3A/3B (inhibition rates less than 20%) at 50 μmol/L. These results indicated the practicability of our strategy to develop cytosine derivatives as DNMT1 inhibitors, and that the length of the linker has a relatively small impact for DNMT1 inhibitory potency. Besides, 5-flurine-substituted cytosine could be acceptable as 206 exhibited comparable DNMT1 inhibition activity. However, analogues of 5-methyl-substituted cytosine, uracil and adenine (i.e., 205, 207 and 208, respectively) caused remarkable decrease of inhibitory activity against DNMT1.
|
|
Table 2 In vitro enzyme inhibitory activities of compounds. |
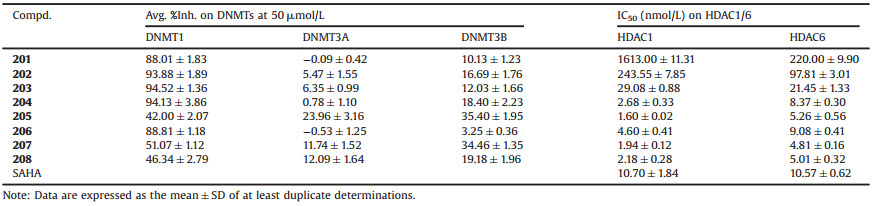
Moreover, our compounds significantly inhibited HDAC1/6 in nanomolar concentrations. For compounds 201–204, the HDAC inhibitory activities improved successively with the increase of linker length of compounds, indicating that the length of linker is critical for HDAC inhibitory potency. Compounds 204–208 presented similar HDAC1/6 inhibition activities, suggesting that different nucleoside bases are tolerable as the cap group for the design of HDAC inhibitors.
Given that compound 204 displayed good DNMT1 and HDAC1/6 inhibitory potency simultaneously, we chose it as a representative compound for further biological evaluations and subsequently tested its DNMT1 inhibitory activity at different concentrations. The results indicated that 204 inhibited DNMT1 with IC50 value of 6.39 μmol/L (Fig. S2 in Supporting information), which is more potent than the reported DNMT inhibitor RG-108 (IC50 = 390 μmol/L) [29] and SGI-1027 (IC50 = 35 μmol/L) [30].
Since 204 exhibited potent DNMT1 and HDAC inhibitory potency in vitro enzymatic inhibition evaluation, we further evaluated its inhibitory activity against DNMT and HDAC at cellular levels. We performed methylation-specific PCR and common PCR evaluations to investigate its effect on the methylation and mRNA level of tumor suppressor gene p16. The results suggested that 204 could effectively inhibited DNMT in U937 cells, increasing the expression of p16 in U937 cells by decreasing the methylation levels in the promoter region of p16 (Fig. 2A). Besides, Western blot assay indicated that compound 204 effectively induced histone H3K9 and H4K8 hyperacetylation in a dose-dependent manner after treating U937 cells for 12 h (Fig. 2B).

|
Download:
|
| Fig. 2. Compound 204 exhibited DNMT and HDAC inhibitory activities at cellular levels. (A) 204 induced the hypomethylation of p16 promoter and increased the expression of p16 mRNA. (B) 204 induced the hyperacetylation of histones H3K9 and H4K8 in U937 cells. p16-U, unmethylated p16 promoter; p16-M, methylated p16 promoter. | |
Furthermore, we measured the effect of 204 on the distribution of cell cycle using flow cytometry assy. The cell cycle profile of U937 after exposure to various concentrations of 204 for 24 h revealed that 204 induced G0/G1 arrest in a dose-dependent manner (Fig. 3). In fact, the G0/G1-phase fraction was increased from 61.02% in the untreated cells to 75.97% in the cells treated with 204 at 12 μmol/L.

|
Download:
|
| Fig. 3. Compound 204 induced U937 cell cycle arrest at G0/G1 phase. | |
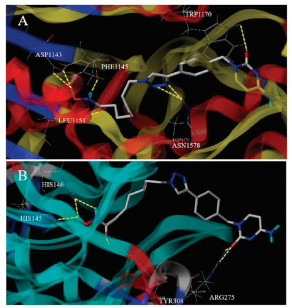
To better understanding the molecular mechanism of compound 204 inhibiting DNMT and HDAC, we explored the binding modes of 204 with the respective enzymes DNMT1 (PDB code: 3SWR) and HDAC2 (PDB code: 4LXZ) using the SYBYL-X 1.3 protocol. As shown in Fig. 4, compound 204 formed 7 hydrogen bonds with DNMT1. The oxygen atom of cytosine group formed one critical hydrogen bond with amino acid residue TRP1170, and the triazole group formed two hydrogen bonds with ASN1578. To our unexpected, the hydroxamic acid group formed four hydrogen bonds with LEU1151, PHE1145 and ASP1143. On the other side, compound 204 formed 6 hydrogen bonds with HDAC2. The cytosine and benzene parts of 204 acting as the cap group occupied the surface area outside the HDAC2 active pocket, as we expected. Besides, the oxygen atom of cytosine group formed two hydrogen bonds with ARG275. The hydroxamic acid group of 204 extended into the active sites through the channel area of HDAC2, forming 4 important hydrogen bonds with HIS145, HIS146 and TYR308. These observations may further confirm the potent inhibitory activity of 204 targeting DNMT1 and HDAC.

|
Download:
|
| Fig. 4. Molecular docking models of compound 204 against (A) DNMT1 and (B) HDAC2. | |
The potent DNMT1 and HDAC1/6 inhibitory activity and good anti-proliferation potency of 204 warranted it as a potential antitumor agent. We then evaluated the metabolic stability of 204 in live microsomes to preliminary estimate its pharmacokinetic properties. As shown in Table 3, 204 exhibited good stability in liver microsomes from three different species (human, mouse and rat). Specifically, 204 is more stable in rat live microsomes with the half life about 6 h, than that in mouse (half life more than 2.4 h) and human live microsomes (half life about 1.7 h). Further investigation about CYP450 isoenzymes inhibition in human live microsomes suggested that 204 could mainly be metabolized by CYP2D6, with IC50 value of 8.7 μmol/L (Table 4). We next sought to explore genotoxicity of 204 by performing Ames screenings. Five mutant strains of Salmonella typhimurium (TA97, TA98, TA100, TA102 and TA1535) were employed. The results (Table 5) showed that compound 204 at concentrations of 50 times that of antiproliferation IC50 values in U937 cells (72 h) displayed mutagenicity in TA98 mutant strain, while approved Histone inhibitor SAHA displayed genotoxicity in TA102 mutant strain.
|
|
Table 3 Metabolic stability of 204 in live microsomes. |

|
|
Table 4 CYP450 inhibition potency of 204. |

|
|
Table 5 Ames evaluation of compound 204. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion, we designed and synthesized a series of hydroxamic acid derivertives of nucleoside bases as dual DNMT and HDAC inhibitors. Representative compound 204 possessed potent inhibitory activity against DNMT1 (IC50 = 6.39 μmol/L), HDAC1 (IC50 = 2.68 nmol/L) and HDAC6 (IC50 = 8.37 nmol/L). Further evaluations indicated 204 could inhibited DNMT and HDAC simultaneously at cellular levels, contributing to the proliferation inhibition of tumor cells. Preliminary studies on metabolic profiles and genotoxicity were also performed. All these tests warrant 204 as an effective DNMT and HDAC dual inhibitor for cancer treatment. Taken together, all these evaluations warranted 204 as a potent DNMT and HDAC dual inhibitor worth further optimization for cancer therapy.
Declaration of competing interestThe authors declared that they have no conflicts of interest to this work. Neither the entire paper nor any part of its content has been published or has been accepted elsewhere.
AcknowledgmentsThe authors would like to thank the financial supports from China Postdoctoral Science Foundation (No. 2018M631825), Shenzhen Development and Reform Committee (No. 2019156), Shenzhen Science, Technology and Innovation Commission (No. JCYJ20180306174248782), Department of Science and Technology of Guangdong Province (No. 2017B030314083) and Shenzhen Bay Laboratory Open Funding (No. SZBL2019062801009).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.004.
| [1] |
S.J. Conway, P.M. Woster, W.J. Greenlee, et al., J. Med. Chem. 59 (2016) 1247-1248. DOI:10.1021/acs.jmedchem.6b00098 |
| [2] |
H.P. Mohammad, O. Barbash, C.L. Creasy, Nat. Med. 25 (2019) 403-418. DOI:10.1038/s41591-019-0376-8 |
| [3] |
D. Morel, D. Jeffery, S. Aspeslagh, et al., Nat. Rev. Clin. Oncol. 17 (2020) 91-107. DOI:10.1038/s41571-019-0267-4 |
| [4] |
P.A. Jones, H. Ohtani, A. Chakravarthy, et al., Nat. Rev. Can. 19 (2019) 151-161. DOI:10.1038/s41568-019-0109-9 |
| [5] |
P. Saravanaraman, M. Selvam, C. Ashok, et al., Biochimie 176 (2020) 85-102. DOI:10.1016/j.biochi.2020.07.004 |
| [6] |
C. Loredana, R. Diego, F.M. Perinelli, et al., Curr. Med. Chem. 27 (2020) 2449-2493. DOI:10.2174/0929867325666181016163110 |
| [7] |
Y. Pan, G. Liu, F. Zhou, et al., Clin. Exp. Med. 18 (2018) 1-14. DOI:10.1007/s10238-017-0467-0 |
| [8] |
A.K.A. Bass, El-Zoghbi M.S., E.S.M. Nageeb, et al., Eur. J. Med. Chem. 209 (2021) 112904. DOI:10.1016/j.ejmech.2020.112904 |
| [9] |
C. Ding, C.Y. Tan, Y.Y. Jiang, et al., Chin. Chem. Lett. 28 (2017) 1220-1227. DOI:10.1016/j.cclet.2017.01.003 |
| [10] |
T. Liu, Y. Wan, Y. Xiao, et al., J. Med. Chem. 63 (2020) 8977-9002. DOI:10.1021/acs.jmedchem.0c00491 |
| [11] |
P. Zhang, X. Wang, X. Liu, et al., Recent Pat. Anticancer Drug Discov. 12 (2017) 16-34. DOI:10.2174/1574892811666161101102842 |
| [12] |
Y. Huang, S. Chen, S. Wu, et al., Acta Pharm. Sin. B 10 (2020) 1294-1308. DOI:10.1016/j.apsb.2019.11.011 |
| [13] |
G. Han, N. Liu, C. Li, et al., J. Med. Chem. 63 (2020) 5341-5359. DOI:10.1021/acs.jmedchem.0c00102 |
| [14] |
Y. Huang, G. Dong, H. Li, et al., J. Med. Chem. 61 (2018) 6056-6074. DOI:10.1021/acs.jmedchem.8b00393 |
| [15] |
G. Dong, W. Chen, X. Wang, et al., J. Med. Chem. 60 (2017) 7965-7983. DOI:10.1021/acs.jmedchem.7b00467 |
| [16] |
S. He, G. Dong, Y. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 3028-3032. DOI:10.1002/anie.201915896 |
| [17] |
K. Fang, G. Dong, Y. Li, et al., ACS Med. Chem. Lett. 9 (2018) 312-317. DOI:10.1021/acsmedchemlett.7b00487 |
| [18] |
W. Chen, G. Dong, Y. Wu, et al., ACS Med. Chem. Lett. 9 (2018) 34-38. DOI:10.1021/acsmedchemlett.7b00414 |
| [19] |
Y. Zhang, L. Xu, A. Li, et al., Biomed. Pharmacother. 110 (2019) 400-408. DOI:10.1016/j.biopha.2018.11.112 |
| [20] |
D. Morel, D. Jeffery, S. Aspeslagh, et al., Nat. Rev. Clin. Oncol. 17 (2020) 91-107. DOI:10.1038/s41571-019-0267-4 |
| [21] |
M.J. Topper, M. Vaz, K.A. Marrone, et al., Nat. Rev. Clin. Oncol. 17 (2020) 75-90. DOI:10.1038/s41571-019-0266-5 |
| [22] |
M.J. Topper, M. Vaz, K.B. Chiappinelli, et al., Cell 171 (2017) 1284-1300. DOI:10.1016/j.cell.2017.10.022 |
| [23] |
D. Tomaselli, A. Lucidi, D. Rotili, et al., Med. Res. Rev. 40 (2020) 190-244. DOI:10.1002/med.21600 |
| [24] |
Z. Yuan, Q. Sun, D. Li, et al., Eur. J. Med. Chem. 134 (2017) 281-292. DOI:10.1016/j.ejmech.2017.04.017 |
| [25] |
Z. Yuan, S. Chen, C. Gao, et al., Bioorg. Chem. 87 (2019) 200-208. DOI:10.1016/j.bioorg.2019.03.027 |
| [26] |
K.J. Falkenberg, R.W. Johnstone, Nat. Rev. Drug Discov. 13 (2014) 673-691. |
| [27] |
J. Roche, P. Bertrand, Eur. J. Med. Chem. 121 (2016) 451-483. |
| [28] |
Y. Luo, H. Li, Int. J. Mol. Sci. 21 (2020) 8828-8853. |
| [29] |
S. Asgatay, C. Champion, G. Marloie, et al., J. Med. Chem. 57 (2014) 421-434. |
| [30] |
S. Valente, Y. Liu, M. Schnekenburger, et al., J. Med. Chem. 57 (2014) 701-713. |