 2021, Vol. 32
2021, Vol. 32 


b College of Chemical Engineering, Nanjing Tech University, Nanjing 211816, China;
c College of Materials Science and Engineering, Nanjing Tech University, Nanjing 211816, China;
d State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China
Coordination polymers (CPs), a class of emerging materials in which metal ions or metal-oxygen clusters are linked by bridged organic ligands via coordination bonds, have been attracted widespread interest owing to their various and tunable crystal structures and physical properties, including luminescence [1, 2], ion conduction [3, 4], dielectrics and ferroelectricity [5], magnetism [6], sorption-separation [7], catalysis [8] and so on. In this context, huge efforts have been devoted to the study of the crystal engineering, aiming to establish the relationship of structure–functionality, by exploring intermolecular interactions, including the long-range interactions, e.g., coordination and H-bond interactions, and the short-range interactions, such as van der Waals force. Although great advances have been made in the last two decades [9-11], there are still many challenges at present and in the future, especially, in the exactly predicting new structures and precisely manipulating the certain functionality of CP-based materials. To address these challenges, abundant energies are still needed to systematically characterize the structures and extensively explore the correlation between the molecular components (linkers and nodes of metal ions or clusters) and the final properties of the resulting structure for new families of functional CPs.
In the area of luminescent materials, room temperature phosphorescent (RTP) CPs exhibit a range of applications in chemical sensing [12], light-emitting diode [13], anti-counterfeiting [14-16] and bioimaging [17] techniques. Theoretically, the heavy atom effect could lead to much improvement of the intersystem crossing process (ISC) rate between different spin states, and finally enhance phosphorescence emission in an organic molecule. It is indeed an efficient strategy to achieve RTP coordination polymers by rational designing the molecular [18-20] and electronic structures of luminescent ligand and choosing the heavy metal ions or heavy metal ion-oxygen clusters as nodes.
As the main group heavy metal, Pb2+ ion shows the coordination numbers from 2 to 12 and diverse coordination geometries, and these structural advantages make Pb2+-based CPs to be one of good candidates for investigating the relationship of phosphorescent nature of a CP and coordination environment of metal ion/carboxylate binding mode, although the Pb2+ ion is harmful to human health. In previous study, we have designed and prepared a series of Pb2+-based CPs and investigated their photophysical properties [21-25]. Unsurprisingly, these CPs we achieved display not only miscellaneous crystal structures, but also diverse luminescent features. To further reveal the relationship of structure and photophysical nature, we go on systematically designing and synthesizing new families of Pb2+-based CPs and exploring their photophysical behaviors.
Herein, we present synthesis, spectroscopic characterization, single crystal structure and luminescent properties of two new Pb2+-CPs, [Pb(FDA)(H2O)] (1) and [NH3(CH3)NH2(CH3)2][Pb4(FDA)5] (2), in which FDA2− is the 2, 5-furandicarboxyle bridging ligand.
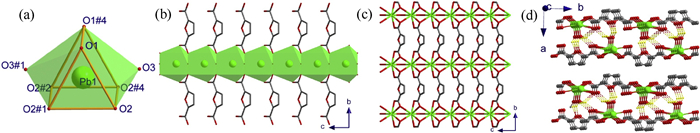
CP 1 crystallizes in orthorhombic space group Pnma, as denoted in Fig. S1a (Supporting information), its asymmetric unit contains one Pb2+ ion which locates at Wyckoff site 4c with a mirror symmetry, one half of deprotonated FDA2− in which the O4 atom occupies Wyckoff site 4c to lead to FDA2− having mirror symmetry, together with one half of disordered H2O molecule which O atom locates at Wyckoff site 4c. The Pb2+ ion is surrounded by eight oxygen atoms from four different carboxylate groups and two disordered water molecules, completing its bicapped triangular prismatic geometry, and the holo-directed coordination sphere of Pb2+ ion has mirror symmetry (Fig. 1a). The Pb-O bond lengths and the O-Pb-O angles are summarized in Table S1 (Supporting information). The geometric parameters in the PbO8 coordination sphere are in agreement with the values reported in other Pb2+ compounds with carboxylates [26]. The bond lengths of d(Pb1–O1#4) = 2.945Å, d(Pb1–O2#2) = d(Pb1–O2#4) = 2.944Å are slight longer, indicating weak coordination bonds between Pb1 and the coordination atoms of O1#4, O2#2, O2#4 [27]. As illustrated in Fig. S1b (Supporting information), each FDA2− ligand links four Pb2+ ions with two carboxylates through bidentate (η1: η2–COO−) binding modes, acting as a μ4−bridge linker. The neighboring bicapped triangular prism are connected together through sharing the planes (O1-O2-O2) into a {PbO5}∞ chain which runs along c-axis (Fig. 1b), and such types of chains extend into a 2D monolayer by weak coordinated bonds between Pb2+ ions and FDA2− ligands as well as between Pb2+ ions and coordinated water molecules (where a water molecule acts as a μ2−bridge linker), and the coordination polymer monolayer is parallel to bc-plane (Fig. 1c). As displayed in Fig. 1d and Fig. S1c (Supporting information), two neighboring monolayers adopt opposite orientations to connect into a bilayer through charge-assisted hydrogen bonds between coordinated water molecules and FDA2− ligands, in which, with respect to one monolayer, another monolayer slides along b- and c-axes c/2 with distances of b/2 and c/2, respectively. The parameters of hydrogen bonds are summarized in Table S3 (Supporting information). Additionally, there are weak van der Waals forces between adjacent bilayers arranged along a-axis, as displayed in Fig. 1d.

|
Download:
|
| Fig. 1. (a) Bicapped triangular prism coordination atmosphere of Pb1, (b) {PbO5}∞ chain running along c-axis, (c) 2D plane constructed by {PbO5}∞ chains and FDA2− ligands viewed along b-axis, (d) H-bonds formed in neighboring monolayers in 1. | |
{kind=link}
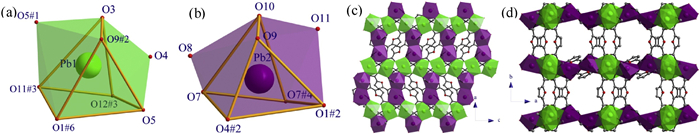
CP 2 also crystallizes in orthorhombic space group Pnma. As illustrated in Fig. S2a (Supporting information), in an asymmetric unit, there are two crystallographically inequivalent Pb2+ ions (labeled as Pb1 and Pb2), one and three halves of deprotonated FDA2− ligands distinguished by the oxygen atoms in the furan rings, as well as one half of disordered dimethylammonium (Me2NH2+) cation and one half of disordered methylammonium (MeNH3+) cation. Both Me2NH2+ and MeNH3+ were produced by the decomposition of DMF solvent during solvothermal reaction process. Both Pb1 and Pb2 are surrounded by eight oxygen atoms to form distorted bicapped triangular prismatic geometry with holo-directed coordination sphere (Figs. 2a and b). The Pb-O bond lengths and the O-Pb-O angles in Pb1 and Pb2 coordination spheres are summarized in Table S2 (Supporting information), all of these bond parameters are comparable to the values in reported Pb-based CPs [26]. Four crystallographically inequivalent FDA2− ligands in 2 show different connectivity. The FDA2− ligand with O6 links five Pb2+ ions (two Pb1 and three Pb2, Fig. S2b in Supporting information), and its carboxylate with O7 bridges two Pb2 through μ2-(η1: η2–COO−) bidentate coordination manner and another one with O4 connects two Pb1 and one Pb2 through μ3-(η2: η2–COO−) bidentate binding modes. The FDA2− ligand with O14 links four Pb2+ ions (two Pb1 and two Pb2, Fig. S2c in Supporting information), and two carboxylates links Pb1 and Pb2 through μ2-(η1: η2–COO−) binding mode. The FDA2− ligands with O2 and O13 show similar connectivity with Pb2+ ions, bridging six Pb2+ ions (four Pb1 and two Pb2 ions, Figs. S2d and e in Supporting information) with two carboxylates through μ3-(η1: η2–COO−) binding modes. As illustrated in Fig. 2c, the neighboring Pb1-type bicapped triangular prisms are arranged into coordination polyhedral chain along c-axis by sharing corners of polyhedra, and the adjacent chains are connected into 2D layer by Pb2-type polyhedral dimer through sharing edges of polyhedra along a-axis, and the layer is parallel to ac-plane. Along b-axis direction, the FDA2− ligands link two neighboring polyhedral layers into pillared-layer framework, which channels are occupied by Me2NH2+ and MeNH3+ cations (Fig. 2d).

|
Download:
|
| Fig. 2. (a, b) Distorted bicapped triangular prismatic coordination geometry of Pb1 (green) and Pb2 (purple), (c) 2D plane being parallel to ac-plane, (d) 3D Hofmann-type networks constructed by 2D layers with FDA2− ligands viewed along c-axis in 2. | |
{kind=link}
In 1 and 2, all Pb2+ ions adopt bicapped triangular prismatic geometry with holo-directed coordination sphere. However, the Pb-O bond lengths are differences in 1 and 2. The Pb–O bond lengths distribute in a broad range and range from 2.435(5) Å to 2.944Å in 1, while in a narrow range in 2 and range from 2.454(9) Å to 2.746(9) Å. In both the CPs, the FDA2− ligands act as both bidentate and bridging ligand. The FDA2− acts as a μ4−bridge linker in 1. However two different coordination modes like bidentate μ2-(η1: η1–COO−) coordination manner and bidentate μ3-(η1: η2–COO−) binding modes are observed in 2. The differences of coordination bond strength and coordination environment of Pb2+ ions result in distinct structure in 1 and 2.
The FT-IR spectra of 1 and 2 are shown in Figs. S3a and b (Supporting information), respectively. The characteristic ν(COO−) and ν(COO−) bands arising from the carboxylate group appear at 1618 cm−1 and 1422 cm−1 in 1 versus 1600 cm−1 and 1366 cm−1 in 2. The differences between ν(COO−) and ν(COO−) is 196 cm−1 in 1 and 234 cm−1 in 2, which are in agreement with the coordination fashion of chelating and bridging carboxylate group [28-30]. PXRD patterns of 1 and 2 are shown in Figs. S4a and b (Supporting information), the good agreement between the experimental and simulated PXRD patterns suggests that both 1 and 2 possess high phase purity. TG plots and their first derivative curves of 1 and 2 are displayed in Figs. S5a and b (Supporting information). In TG plot of 1, the abrupt weight loss starts at ca. 240 ℃, related to the losing coordinated water molecules, as well as the disintegration of the framework. The TG and DTA analysis for 2 reveals that the abrupt weight loss starts at 249 ℃, corresponding to the decomposition of the charge balancing cations of MeNH3+ and Me2NH2+, and the anionic framework disintegrates when the temperature is further elevated.
The solid-state diffuse reflection spectra at ambient temperature are displayed in Figs. S6a and b (Supporting information) for 1, 2 and H2FDA. All absorption bands of H2FDA fall within the ultraviolet spectral region, with three shoulders around 263, 295 and 320 nm, attributed to the π→π* and n→π* electron transition in the furan rings and carboxyl groups. Different from the absorption profile of H2FDA, the broad absorption bands span from 200 nm to 450 nm in 1 and 2, and the low-energy shoulder redshift occurs and becomes visible with the maximum at ~336 nm in 1 versus ~383 nm in 2 owing to perturbation of Pb2+ ions to the energy levels of FDA2– ligands, and it is similar to that in H2FDA, the intensity of the low-energy shoulder band is less than that of the bands at high energy side in both CPs. As shown in Fig. S3c (Supporting information), the optical band gaps (Eg) of 1 and 2 are estimated as 3.92 and 3.72 eV from the Tauc plots, in agreement with the colorless sample under daylight. Under UV light (λ= 330–380 nm), 1 shows yellow-green, while 2 pale yellow color (Figs. S7a-d in Supporting information).
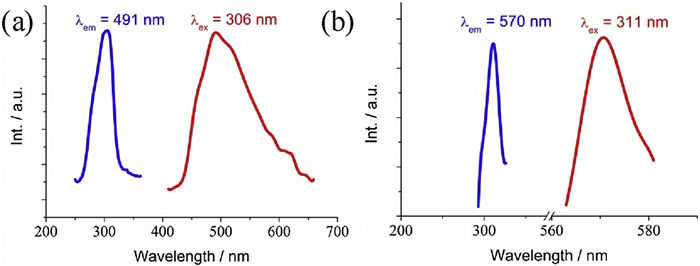
The luminescent properties of 1 and 2 together with H2FDA were studied at ambient condition. In Fig. S6d (Supporting information), the emission spectrum of H2FDA shows two intense emission bands centered around 375 nm and 383 nm under the irradiation of λex = 301 nm due to the transitions n→π* or π→π*. As denoted in Figs. 3a and b, the emission spectrum presents a band with the maximum at 491 nm upon excitation at 301 nm for 1, a band with the maximum at 570 nm upon UV irradiation at 311 nm for 2, and all of these emission bands are assigned to electron transition within FDA2− ligands. The luminescent color of 1, 2 and H2FDA are consistent with the corresponding CIE indexes (Fig. S7e in Supporting information). It is worth mentioning that, by comparison of H2FDA, the emission bands in 1 and 2 pronouncedly redshift, indicating that the coordination of Pb2+ ions strongly affects the electron energy levels of FDA2− ligands. It is also noted that, regarding 1, the emission band in 2 shows larger redshifts even through the Pb2+ ions in the crystal structures of 1 and 2 show similar coordination sphere. To inspect the coordination environments of Pb2+ ions reveals that most of Pb–O bond lengths in 1 and 2 are comparable to each other, but three Pb–O bond lengths are longer in 1. The different strengths of coordination bonds of Pb2+ ion and FDA2− ligand results in the distinct perturbation of Pb2+ ion to the electron energy level of FDA2− ligand.

|
Download:
|
| Fig. 3. Solid luminescence spectrum of (a) 1 and (b) 2. | |
{kind=link}
The luminescence decay curves of H2FDA, 1 and 2 under ambient conditions are shown in Figs. S8a-c (Supporting information), and the luminescence lifetime (τ) was gained by fitting the curve using an single exponential decay function, and the best fits give the τ = 0.62 ms for 1 versus 1.69 ms for 2, falling within the millisecond range and showing the phosphorescence character. However, the τ value of H2FDA is only 1.7 ns, indicating the typical organic fluorescence character. The quantum yields (QY) in 1 and 2 were estimated as 14.9% and 15.7%, respectively, under ambient condition. The long lifetimes and high quantum yields of 1 and 2 demonstrate that the heavy atom effect of Pb2+ really improves the intersystem crossing efficiency between excited singlet (S1) and triplet states (T1) of the FDA2− ligand.
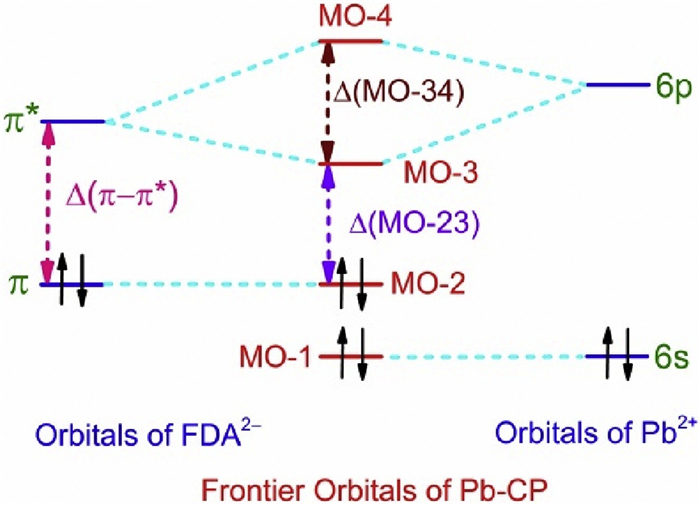
H2FDA emits fluorescence, while 1 and 2 show the ligand-centered phosphoresce owing to the heavy atom effect of Pb2+ ions, moreover, the optical bandgap increases in the sequence of 2, 1 and H2FDA, and this tread of change is in good agreement with the variation of the wavelength at the maximum intensity of emission band in 2 (λem = 570 nm), 1 (λem = 491 nm) and H2FDA (λem = 375 nm). The finding is understandable using the qualitative frontier orbital diagram analysis of a Pb-CP. For simplicity, we suppose, the frontier orbitals of a Pb-CP consist of the occupied π orbital with two electrons and unoccupied π* orbital in FDA2− ligand as well as the 6 s orbital with a lone pair of electrons and the unoccupied 6 p orbitals in Pb2+ ion. It is well known that the Pb4+ ion shows strong oxidbillity as reduced to Pb2+ species, while the Pb metal has strong reducibility as oxidized to Pb2+ species, and these facts demonstrate that the Pb2+ species is hard either oxidized or reduced. It is reasonable to assume that the energy levels of the 6 s and 6 p orbitals in the Pb2+ ion are respectively lower and higher than the energy levels of the π and π* orbitals in FDA2− ligand. On the basis of molecular orbital theory, the molecular orbital consists of the linear combination of atomic orbitals with the same symmetry and similar energy level. On the other hand, the s and p orbitals of atom show different symmetry, and the π orbital are comprised of p orbitals of atom. Accordingly, the energy level diagram of frontier orbitals in a Pb-CP and the relationship of the frontier orbitals in a Pb-CP with the atomic orbitals in Pb2+ ion and FDA2− ligand may be approximately illustrated in Fig. 4, in which the symbols of Δ(π–π*), Δ(MO-23) and Δ(MO-34) represent the energy differences of HOMO and LUMO in a FDA2− ligand and a Pb-CP, the splitting energy between the molecular orbitals of MO-2 and MO-3, as well as between molecular orbitals of MO-3 and MO-4. It is worth noting that the splitting energy Δ(MO-34) is strongly dependent on the orbital interaction between the 6 p orbitals in the Pb2+ ion and the π* orbital in the FDA2− ligand, which is relevant to the strength of Pb-O coordination bonds. Undoubtedly, the energy difference of HOMO and LUMO in the FDA2− ligand is bigger than that in a Pb-CP, and the stronger Pb-O bonds in the coordination sphere of Pb2+ ion should give the bigger splitting energy of Δ(MO-34), leading to smaller energy level difference of Δ(MO-23). The fact of the stronger Pb-O bonds in 2 than those in 1 responds to the redshift of the λem in 2 regarding the λem in 1.

|
Download:
|
| Fig. 4. Schematic illustration of the energy levels of the frontier orbitals in Pb2+ ion, FDA2− ligand and Pb-CP. | |
{kind=link}
In summary, the crystal structure and photophysical property have been investigated for two RTP Pb2+-based CPs. The Pb2+ ions show bicapped triangle prism coordination geometry and these coordinated triangle prisms are connected through FDA2− ligands into a bilayer coordination polymer in 1, while a 3D pillared-layer framework in 2. The distinct connectivity of FDA2− ligands in 1 and 2 leads to their different Pb2+-ligand coordination interactions. Both 1 and 2 emit ligand-centered RTP owing to the heavy atom effect of Pb2+ ion. The qualitative analysis for frontier orbitals demonstrates that the emission band red-shift in 2 regarding 1 arises from the different coordination interactions between Pb2+ ions and ligands. Our study clearly demonstrates the relationship of ligand-centered photophysical property of a CP and metal-ion-ligand coordination interactions, offering a way for insight understanding into the structure–luminescence relationship.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThe authors thank the National Natural Science Foundation of China (No. 21901116).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.015.
| [1] |
Y. Pointel, Y. Suffren, C. Daiguebonne, et al., Inorg. Chem. 59 (2020) 10673-10687. DOI:10.1021/acs.inorgchem.0c01136 |
| [2] |
Y. Yang, X. Yang, X. Fang, K.Z. Wang, D. Yan, Adv. Sci. 5 (2018) 1801187. DOI:10.1002/advs.201801187 |
| [3] |
S. Kim, B. Joarder, J.A. Hurd, et al., J. Am. Chem. Soc. 140 (2018) 1077-1082. DOI:10.1021/jacs.7b11364 |
| [4] |
R. Moi, A. Ghorai, S. Banerjee, K. Biradha, Cryst. Growth Des. 20 (2020) 5557-5563. DOI:10.1021/acs.cgd.0c00732 |
| [5] |
L.H. Chen, J.B. Guo, X. Wang, et al., Adv. Mater. 29 (2017) 1702512. DOI:10.1002/adma.201702512 |
| [6] |
S.E. Henkelis, D.L. Huber, D.J. Vogel, J.M. Rimsza, T.M. Nenoff, ACS Appl. Mater. Interfaces 12 (2020) 19504-19510. DOI:10.1021/acsami.0c01813 |
| [7] |
S. Noro, T. Nakamura, NPG Asia Mater. 9 (2017) e433. DOI:10.1038/am.2017.165 |
| [8] |
X. Liu, T. Yue, K. Qi, et al., Chin. Chem. Lett. 31 (2020) 2189-2201. DOI:10.1016/j.cclet.2019.12.009 |
| [9] |
E. Li, K. Jie, M. Liu, et al., Chem. Soc. Rev. 49 (2020) 1517-1544. DOI:10.1039/C9CS00098D |
| [10] |
W.P. Lustig, S. Mukherjee, N.D. Rudd, et al., Chem. Soc. Rev. 46 (2017) 3242-3285. DOI:10.1039/C6CS00930A |
| [11] |
B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn, J. Gascon, Coord. Chem. Rev. 307 (2016) 147-187. DOI:10.1016/j.ccr.2015.06.008 |
| [12] |
R. Martín-Rodríguez, F. Aguado, M.D. Alba, R. Valiente, A.C. Perdigo'n, ACS Appl. Mater. Interfaces 11 (2019) 7559-7565. DOI:10.1021/acsami.8b20030 |
| [13] |
Z. You, H. Li, L. Zhang, et al., J. Phys. Chem. C 121 (2017) 23072-23079. DOI:10.1021/acs.jpcc.7b07627 |
| [14] |
B. Zhou, Q. Zhao, L. Tang, D. Yan, Chem. Commun. (Camb.) 56 (2020) 7698-7701. DOI:10.1039/D0CC02730H |
| [15] |
B. Zhou, D. Yan, Angew. Chem. Int. Ed. 58 (2019) 15128-15135. DOI:10.1002/anie.201909760 |
| [16] |
R. Gao, D. Yan, D.G. Evans, X. Duan, Nano Res. 10 (2017) 3606-3617. DOI:10.1007/s12274-017-1571-x |
| [17] |
K.S. Butler, C.J. Pearce, E.A. Nail, G. Vincent, D.F.S. Gallis, ACS Appl. Mater. Interfaces 12 (2020) 31217-31224. DOI:10.1021/acsami.0c07835 |
| [18] |
B. Zhou, D. Yan, Sci. China Chem. 63 (2020) 423-425. DOI:10.1007/s11426-019-9691-2 |
| [19] |
B. Zhou, D. Yan, Adv. Funct. Mater. 29 (2019) 1807599. DOI:10.1002/adfm.201807599 |
| [20] |
X. Yang, D. Yan, Adv. Opt. Mater. 4 (2016) 897-905. DOI:10.1002/adom.201500666 |
| [21] |
X.S. Gao, H.J. Dai, M.J. Ding, W.B. Pei, X.M. Ren, Inorg. Chem. 58 (2019) 6772-6780. DOI:10.1021/acs.inorgchem.9b00215 |
| [22] |
X.S. Wu, Y.X. Wang, S.Q. Li, et al., Dalton Trans. 47 (2018) 14636-14643. DOI:10.1039/C8DT03589J |
| [23] |
J.Y. He, Z.R. Deng, X. Liu, et al., Dalton Trans. 47 (2018) 9334-9340. DOI:10.1039/C8DT01951G |
| [24] |
X. Liu, L. Zhai, W.W. Zhang, et al., Dalton Trans. 46 (2017) 7953-7959. DOI:10.1039/C7DT01626C |
| [25] |
X.S. Wu, Y.R. Tang, J.L. Liu, L. Wang, X.M. Ren, Dalton Trans. 48 (2019) 13841-13849. DOI:10.1039/C9DT02928A |
| [26] |
R.L. Davidovich, V. Stavila, D.V. Marinin, E.I. Voit, K.H. Whitmire, Coord. Chem. Rev. 253 (2009) 1316-1352. DOI:10.1016/j.ccr.2008.09.003 |
| [27] |
A. Bondi, J. Phys. Chem. 68 (1964) 441-451. DOI:10.1021/j100785a001 |
| [28] |
G.B. Deacon, R.J. Phillips, Coord. Chem. Rev. 33 (1980) 227-250. DOI:10.1016/S0010-8545(00)80455-5 |
| [29] |
L.J. Zhang, J.Q. Xu, Z. Shi, W. Xu, T.G. Wang, Dalton Trans. (2003) 1148-1152. |
| [30] |
M. Tabatabaee, S. Amjad, S. Tabatabaei, K. Molcanov, Synth. React. Inorg. Met. 944 (2014) 507-513. |