 2021, Vol. 32
2021, Vol. 32 


b College of Chemical Science and Technology, Yunnan University, Kunming 650091, China
Hydrogen sulfide (H2S) is the third gas signal molecule (gasotransmitter) after carbon monoxide (CO) and nitric oxide (NO), and is associated with a wide range of physiological functions in the human body [1-4]. Endogenous H2S is enzymically synthesized by mammalian tissues via three enzymes, including cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST) [5, 6]. Abnormal concentrations of H2S causes many human diseases, such as Alzheimer's, liver cirrhosis, diabetes and gastric mucosal injury [7-10]. In particular, H2S concentration is closely related to colorectal cancer [11], which is a multifactorial disease caused by lifestyle, genetics and environmental factors [12]. As colon cancer cells express more CBS than normal cells, they can produce and release more H2S in the tumor microenvironment [13]. Compared with traditional detection methods, visual detection of H2S concentration in biological systems is of great significance. Compared with other detection methods, the use of fluorescent molecular probes to detect objects in organisms has irreplaceable advantages in biological imaging, and a lot of related work has been achieved [14-18]. As a result, numerous fluorescent probes for H2S detection have been developed in recent years [19-24]. Li et al. reported fluorescent probe DBT for visual detection of H2S in colorectal cancer cells, which is composed of triphenylamine, BODIPY and 2, 4-dinitrobenzenesulfonyl group [25]. In 2020, Muthusamy et al. designed a fluorophore Lyso-Rh-S-DNP by orderly binding of 2, 4-dinitrophenyl and salicylaldehyde to a rhodamine backbone, which allowed quantitative H2S detection in organisms [26]. In our previous work, we developed a fluorescent H2S probe to observe H2S in living cells and Caenorhabditis elegans (C. elegans) [27]. The results showed that fluorescent detection can provide simple and convenient methods for H2S detection in cells. Therefore, the use of H2S as an initiator for drug release is feasible for the treatment of tumors, especially colorectal cancer.
The theranostic system, which integrates cancer signature species detection and the release of therapeutic drug, has attracted great attention in recent years [28]. The most basic fluorescent theranostic system is composed of three parts: fluorophore, masked anticancer active drug, and connecting linker, which can be cleaved in response to a particular over expressed species in the cancer cells [29]. Based on this design concept, a variety of studies have been reported [30-32]. In recently, Zheng et al. reported a nitroreductase-activatable theranostic molecule, which can be used in biological imaging and therapy cancer cell [33]. However, compared with the application of various fluorophores and anticancer drugs, there is a lack of innovation in the exploration of drug release triggers.
Compared with one-photon fluorescence imaging technology, two-photon fluorescence imaging has incomparable advantages in the detection of active species [34-36]. As the excitation wavelength is located in the near infrared region (700-900 nm), it causes less photodamage to biological samples, increases tissue penetration and effectively reduces interference of biological background [37]. Therefore, the development of a two-photon fluorescent theranostic system with novel H2S-triggered drug release mechanisms is desirable.
In this work, we reported the design and synthesis of a new two-photon H2S-triggered ratiometric fluorescent theranostic prodrug (compound 1), for in vivo imaging. Scheme 1 shows that compound 1 is composed of three functional parts: naphthalimide as the ratiometric fluorophore and two-photon signal reporter, with a disulfide bond as the cleavable linker, which can be triggered by H2S, and 7-ethyl-10-hydroxycamptothecin (SN-38) as the anticancer drug. As expected, the disulfide bond in compound 1 underwent cleavage in the presence of H2S, promoting the release of free active drug SN-38, accompanied by a red-shifted fluorescence signal from 460 nm to 545 nm. The bio-imaging of exogenous and endogenous H2S in living cells by compound 1 was successfully applied under both one-photon and two-photon excitation. An effective drug release in situ tumor, as well as fluorescence imaging were verified when compound 1 was injected into nude mice. It showed that compound 1 is a promising therapeutic compound for future cancer therapy.

|
Download:
|
| Scheme 1. Proposed SN-38 release and fluorescent variation mechanism of the compound 1 with H2S. | |
{kind=link}
The synthetic route for compound 1 is outlined in Scheme S1 (Supporting information). The developed ratiometric two-photon theranostic prodrug was prepared using prototype intramolecular charge transfer (ICT) fluorophore 1, 8-naphthalimide, which exhibits excellent spectral properties, such as high absorption coefficient and high quantum yield in the visible region, high photostability, large Stokes' shift and significant two-photon cross section. 4-Nitro-1, 8-naphthalic anhydride (compound 5) was reacted with lysosomal targeting group 4-(2-aminoethyl)morpholine in ethanol producing N-(2-morpholinoethyl)-4-nitro-1, 8-naphthalimide (compound 4) in 73% yield. N-(2-Morpholinoethyl)-4-amino-1, 8-naphthalimide (compound 3) was prepared via the reaction of compound 4 with SnCl2·2H2O in the presence of concentrated hydrochloric acid in MeOH, where the nitro group was reduced to an amino group. In the following reaction, 3, 3-dithiodipropionic chloride was prepared by reacting 3, 3-dithiodipropionic acid with SOCl2 in anhydrous dichloromethane under an argon atmosphere. Then, compound 3 was reacted with 3, 3-dithiodipropionic chloride in the presence of 4-dimethylaminopyridine (DMAP), followed by treatment with sodium bicarbonate and dilute hydrochloric acid solution producing compound 2. In the case of the reaction involving the introduction of anticancer drugs, highly toxic phosgene solution or triphosgene was avoided, instead less toxic EDCI (1-ethyl-(3-dimethylaminopropyl)carbodiimide hydrochloride) and DMAP reaction were employed to connect compound 2 and SN-38 in order to produce target compound 1. The detailed procedures and characterization of the new compounds are described in the Supporting information.
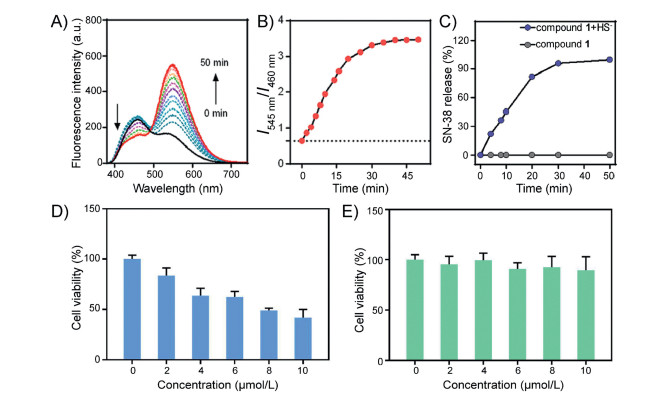
To evaluate whether compound 1 could activate in the presence of H2S, we first tested the absorption and fluorescent responses of compound 1 in the absence or presence of NaHS (a commonly used H2S source) in a mixed solution of DMSO/PBS (1/1, v/v, pH 7.4) at ambient temperature (Fig. 1). Owing to the typical ICT structure of naphthalimide, compound 1 exhibited an absorption band centered at 370 nm and a relatively weak fluorescence band centered at 460 nm. When excess HS- (400 μmol/L) was added to compound 1 (20 μmol/L) in DMSO/PBS (1/1, v/v, pH 7.4), the absorption band at 370 nm decreased and a new band with a shoulder at 430 nm appeared (Fig. 1A), accompanied by the visible change of the colorless solution to jade-green (Fig. S1 in Supporting information). Additionally, a change in the fluorescence spectrum was observed when excited at 370 nm, in which the emission intensity at 460 nm decreased and that at 545 nm dramatically increased (Fig. 1B). Thus, the solution revealed significant ratiometric fluorescence change from blue to yellow-green (Fig. S1). The absorption and fluorescence titration spectra of compound 1 with H2S were consistent with our predicted detection mechanism (Scheme 1). After reacting with H2S, the disulfide bond (S-S) of compound 1 was cleaved and BHNP-NH2 fluorophore was released, where the amide group with electron-withdrawing substituents transformed into the electron-donating amino group resulting in enhanced ICT effect of the naphthalimide fluorophore, leading to the remarkable red shift of both absorption (from 370 nm to 430 nm) (Fig. 1C) and fluorescence spectra (from 460 nm to 545 nm) (Fig. 1D).

|
Download:
|
| Fig. 1. Absorption (A) and absorption titration spectra (C), fluorescence (B) and fluorescence titration spectra (D) of compound 1 (20 μmol/L) with or without HS– (0.4 mmol/L) and incubation for 50 min in a mixture solution of DMSO/PBS (1/1, v/v, pH 7.4), λex=370 nm. (E) Time-dependent fluorescence ratio (I545 nm/I460 nm) responses of compound 1 (20 μmol/L) toward HS– (200 μmol/L), Cys (200 μmol/L), and GSH (200 μmol/L) in PBS (pH 7.4), λex=370 nm, (F) Fluorescence intensity ratio changes with the concentration of HS–. Note: the detection limit was calculated based on the fluorescence titration. The limit of detection for HS– is 0.72 μmol/L. | |
{kind=link}
High selectivity to the test object is an essential condition for a fluorescent probe with excellent performance. In order to verify the selectivity of the compound 1 toward HS–, we tested the absorption and fluorescent responses (Fig. S2 in Supporting information) of compound 1 (20 μmol/L) in the presence of various anions (200 μmol/L) in a mixture solution of DMSO/PBS (1/1, v/v, pH 7.4). The results showed that no obvious absorption and fluorescent responses were found by the addition of various anions except for HS– to the solution of compound 1.
More importantly, compound 1 also showed good selectivity for HS– comparing to the sulfur-containing amino acids, such as Cys and GSH, which can induce disulfide bond breakage (Fig. 1E). In order to test whether compound 1 can be quantitatively detected for H2S, we diagramed the fluorescence intensity ratio of two peaks (I545 nm/I460 nm) as a function of the H2S concentrations. The fluorescence intensity ratio of two peaks (I545 nm/I460 nm) exhibited a good linear relationship with the H2S concentration and the detection limit was 0.72 μmol/L (Fig. 1F).
The proposed reaction mechanism is shown in Scheme 1, which is similar to our previous work. We previous reported a cyanine-based colorimetric and fluorescence probe for H2S detection in vivo [27], as well as lysosome-targeted fluorescent H2S probe for H2S visualization in living cells and tracing accumulated lysosome [37]. Both approaches employed the same disulfide bond linker as compound 1, and the proposed mechanism involved disulfide bond cleavage triggered by H2S. After disulfide bond cleavage, the fragments underwent intramolecular cyclization, releasing five-member ring compound 1, 2-dithiolan-3-one and naphthalimide fluorophore (BHNP-NH2) as well as SN-38 anticancer drugs. To verify this mechanism, the absorbance and fluorescence spectra of BHNP-NH2 (compound 3) were compared with that of compounds 1 after H2S treatment (Fig. S1). After the addition of H2S, compound 1 produced a shoulder peak in the absorption spectrum located at 400-500 nm and a new fluorescence emission peak at 500-600 nm, which was consistent with that of compound 3. After compound 1 reacted with H2S, the solution color changed from colorless to light yellow-green, and the fluorescence emission color changed from blue to yellow-green, indicating that compound 1 released free compound 3 through disulfide bond cleavage triggered by H2S. In addition, the proposed mechanism was verified by ESI-MS analyses. Upon addition of 40 equiv. H2S to compound 1, four distinct mass spectral debris signal peaks located at m/z 325.0645 (corresponding to BHNP-NH2), 349.1550 (corresponding to BHNP-NH2+Na+), 365.1165 (corresponding to BHNP-NH2+K+) and 393.1441 (corresponding to SN-38+H+) were observed by high resolution MS spectroscopy (Fig. S3 in Supporting information). This clearly showed that after compound 1 was triggered by H2S, it effectively released anticancer drug SN-38 and the fluorescence signal reporter group compound 3 via cleavage of the disulfide bond.
In order to further confirm that SN-38 drug release was triggered by H2S, we conducted a time-dependent experiment in which the change of fluorescence ratio I545 nm/I460 nm was monitored. As shown in Figs. 2A and B, upon addition of HS– (400 μmol/L) to compound 1 (20 μmol/L) in a solution of DMSO/PBS (1/1, v/v, pH 7.4), within 50 min the fluorescence emission intensity at 460 nm displayed a gradual decrease, and the fluorescence emission intensity at 545 nm a gradual increase. This was in accordance with the fluorescence ratio I545 nm/I460 nm changed from 0.7 to maximum of 3.5. To verify that SN-38 drug release process coincided with the fluorescence change of compound 1, the SN-38 release process was analyzed by RP (reverse phase)-HPLC chromatograms, where the drug release value of compound 1 exceeded 90% within 50 min (Fig. 2C).

|
Download:
|
| Fig. 2. Time-dependent of (A) fluorescence spectra and (B) I545 nm/I460 nm (0–50 min) for compound 1 (20 μmol/L) in a mixture solution of DMSO/PBS (1/1, v/v, pH 7.4) with HS– (400 μmol/L), λex=370 nm. (C) SN-38 released from compound 1 (20 μmol/L) as a function of time in the presence and absence of HS– (400 μmol/L). SN-38 in RP (reverse phase)-HPLC chromatograms was detected by UV absorption using 370 nm as the monitored wavelength. Cell viability of compound 1 and reference compound 2 at different concentrations. HCT116 cells (colon cancer cells) were incubated with various concentrations (0–10 μmol/L) compound 1 (D) and reference compound 2 (E) for 24 h. Data are shown as mean±S.D., n=3. | |
{kind=link}
Based on the time-dependent release of SN-38, we conducted cytotoxicity tests using compound 1 and reference compound 2 (details in Supporting information) in HCT116 cells in vitro. Even compound 2 has the similar structure with compound 1, it can not release SN-38 after breaking disulfide bond. The results are summarized in Figs. 2D and E. When HCT116 cells (colon cancer cells) were incubated with various concentrations (0-10 μmol/L) of compound 1 (Fig. 2D), it was found that the cell activity showed a gradual but significant decrease. After the addition of 10 μmol/L compound 1, the cell viability decreased by 50%. However, little change in the cell viability was observed for reference compound 2 (Fig. 2E). These results were consistent with our hypothesis, and suggested that H2S in colon cancer cells could induce compound 1 to release anticancer drug SN-38.
In order to verify that compound 1 could locate endogenous and exogenous H2S in living cells cumulating in drug release, we performed fluorescence imaging experiments. As shown in Fig. 3, HCT116 and A549 cells exhibited a more obvious blue fluorescence and less obvious green fluorescence after incubation with compound 1 for 30 min. SAM (S-adenosyl-l-methionine) acts as the promoter of cellular endogenous H2S. Hence, when HCT116 cells were pretreated with 3 mmol/L SAM for 1 h, followed by incubation with compound 1 (10 μmol/L) for 30 min, the blue fluorescence decreased slightly, and green fluorescence increased significantly, where the ratio of green and blue fluorescence displayed an obvious increase. The opposite of SAM is AOAA (aminooxyacetic acid), which is an inhibitor of cellular endogenous H2S. When HCT116 cells were pretreated with 1 nmol/L AOAA for 1 h, followed by incubation with compound 1 (10 μmol/L) for 30 min, the blue fluorescence showed significant enhancement, while the green fluorescence only slightly weakened, and the ratio of green and blue fluorescence was significantly reduced.

|
Download:
|
| Fig. 3. Identification of colon cancer cells by confocal microscopy imaging in dual-color modality. (a1-a4) HCT116 cells incubated with compound 1 (10 μmol/L) for 30 min. (b1-b4) HCT116 cells pretreated with 3 mmol/L SAM for 1 h, followed by incubation with compound 1 (10 μmol/L) for 30 min. (c1-c4) HCT116 cells pretreated with AOAA (1 mmol/L) for 1 h were loaded with compound 1 (10 μmol/L) for 30 min. (d1-d4) A549 cells incubated with compound 1 (10 μmol/L) for 30 min. λex=400 nm, blue channel at 410–500 nm, green channel at 510–650 nm; ratio images generated from green channel to blue channel. | |
{kind=link}
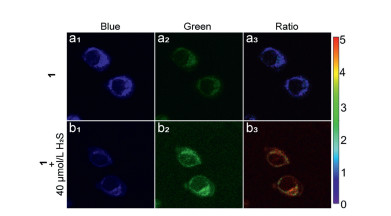
Based on single-photon fluorescence imaging, two-photon fluorescence imaging was performed. As shown in Fig. 4, when excited at 740 nm, compound 1 (10 μmol/L) in HeLa cells displayed strong blue fluorescence and weaker green fluorescence. When HeLa cells pretreated with H2S (40 μmol/L) for 30 min, followed by incubation with compound 1 (10 μmol/L) for 30 min, the blue fluorescence decreased and green fluorescence increased, and the ratio of green and blue fluorescence showing an obvious increase. Hence, compound 1 had good capability of two-photon imaging for H2S detection and drug release.

|
Download:
|
| Fig. 4. Two-photon image of compound 1 (10 μmol/L). HeLa cells incubated with compound 1 for 30 min (a1-a3). HeLa cells pretreated with H2S (40 μmol/L) for 30 min, followed by incubation with compound 1 for 30 min (b1-b3), λex=740 nm. Ratio images generated from green channel to blue channel. | |
{kind=link}
Determining if compound 1 could release SN-38 in C. elegans under H2S was also examined through fluorescence imaging experiments. All the animal studies were carried out in the Yunnan University, with the approval number of yuncare 20200353.C. elegans was pretreated with different concentrations of H2S for 2 h, and then incubated with compound 1 (20 μmol/L) for 2 h. As shown in Fig. 5, C. elegans shows a strong blue and weak green fluorescence in the absence of H2S. In contrast, as the concentration of pretreated H2S increases, the blue fluorescence of C. elegans gradually decreases and green fluorescence gradually increases in a dose-dependent manner. This result was consistent with our previous hypothesis that compound 1 may release SN-38 in the presence of H2S.

|
Download:
|
| Fig. 5. Bright field images and fluorescence images of C. elegans. (a1-a4) C. elegans incubated with compound 1 (20 μmol/L) for 2 h. (b1-b4)C. elegans pretrated with H2S (100 μmol/L) for 2 h, followed by incubated with compound 1 (20 μmol/L) for 2 h. (c1-c4)C. elegans pretrated with H2S (300 μmol/L) for 2 h, followed by incubated with compound 1 (20 μmol/L) for 2 h. Scale bar: 20 μm. | |
{kind=link}
The drug release of compound 1 in A549 cell tumor-bearing mice was investigated via in vivo fluorescence imaging. In group 1, tumor-bearing mice were in situ injected with compound 1 (5 mg/kg) at the tumor site. In group 2, tumor-bearing mice were first injected with NaHS (5 mg/kg) and after 30 min injected with compound 1 (5 mg/kg) at the tumor site. After 3 h, the tumor and other major organs of both tested mice were dissected and analyzed by fluorescence imaging. As shown in Fig. 6, the tumor sites of both groups showed obvious fluorescence compared to the other major organs, such as the heart, liver, spleen, lung and kidney. In comparison, the rat in group 1 showed that the fluorescence intensity at the tumor site was considerably greater than that in group 2, which possessed exogenous NaHS. These results indicate that endogenous H2S could cause bond cleavage and release of the fluorescence moiety. Furthermore, addition of exogenous H2S increased the release of fluorescent indicators BHNP-NH2 and therapeutic drugs SN-38, which led to stronger fluorescence signals at the tumor site. Therefore, compound 1 could response to endogenous and exogenous H2S promoting the release of therapeutic drugs and fluorescent emission.

|
Download:
|
| Fig. 6. Fluorescence images of the internal organs after anatomy A549 cell tumor-bearing mice after in situ injection of compound 1 (5 mg/kg) in tumor site for 3 h (up), first in situ injection of NaHS (5 mg/kg) in tumor site for 0.5 h, then in situ injection of compound 1 (5 mg/kg) in tumor site for 3 h (down). | |
{kind=link}
In summary, through the conjugation of anticancer drug SN-38 and naphthalimide fluorophores via a disulfide bond, we developed a novel H2S-responsive ratiometric fluorescent theranostics prodrug (compound 1). The obtained results showed that it was able to react with endogenous and exogenous H2S, accompanied by release of anticancer drug SN-38 and significantly inhibited the activity of colorectal cancer cell HCT116. More importantly, compound 1 was applied in the bio-imaging of endogenous and exogenous H2S in cancer cells, C. elegans, and tumor-bearing mice, under one-photon or two-photon excitation. Compound 1 provided a promising platform for specific tumor-activatable drug delivery system, which can be applied in future cancer treatment and therapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank for the support of the National Natural Science Foundation of China (Nos. 21867019, 22067021, 22067019) and the "Youth Talent of WanRen Project" Foundation of Yunnan Province of China. J.F. Zhang acknowledges the "LianDa Scholar Project" of Yunnan Normal University.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.065.
| [1] |
L. Li, P. Rose, P.K. Moore, Annu. Rev. Pharmacol. Toxicol. 51 (2011) 169-187. DOI:10.1146/annurev-pharmtox-010510-100505 |
| [2] |
Łowicka E., J. Bełtowski, Pharmacol. Rep. 59 (2007) 4-24. |
| [3] |
N.L. Kanagy, C. Szabo, A. Papapetropoulos, Am. J. Physiol.: Cell Ph. 312 (2017) C537-C549. DOI:10.1152/ajpcell.00329.2016 |
| [4] |
S.C. Kang, E.H. Sohn, S.R. Lee, Oxid. Med. Cell. Longev. 2020 (2020) 4105382. |
| [5] |
N. Bazhanov, M. Ansar, T. Ivanciuc, R.P. Garofalo, A. Casola, Am. J. Respir. Cell Mol. Biol. 57 (2017) 403-410. DOI:10.1165/rcmb.2017-0114TR |
| [6] |
N. Dilek, A. Papapetropoulos, T. Toliver-Kinsky, C. Szabo, Pharmacol. Res. 161 (2020) 105119. |
| [7] |
M.A. Aminzadeh, N.D. Vaziri, Nephrol. Dial. Transplant. 27 (2012) 498-504. DOI:10.1093/ndt/gfr560 |
| [8] |
B.D. Paul, S.H. Snyder, Biochem. Pharmacol. 149 (2018) 101-109. DOI:10.1016/j.bcp.2017.11.019 |
| [9] |
Coavoy-Sánchez S.A., S.K. Costa, M.N. Muscará, Br. J. Pharmacol. 177 (2020) 857-865. DOI:10.1111/bph.14699 |
| [10] |
M.R. Hellmich, C. Coletta, C. Chao, C. Szabo, Antioxid. Redox Sign. 22 (2015) 424-448. DOI:10.1089/ars.2014.5933 |
| [11] |
G. Oláh, K. Módis, G. Törö, et al., Biochem. Pharmacol. 149 (2018) 186-204. DOI:10.1016/j.bcp.2017.10.011 |
| [12] |
V. Aran, A.P. Victorino, L.C. Thuler, C.G. Ferreira, Clin. Colorectal. Canc. 15 (2016) 195-203. DOI:10.1016/j.clcc.2016.02.008 |
| [13] |
S. Chen, T. Yue, Z. Huang, et al., Cancer Lett. 466 (2019) 49-60. DOI:10.1016/j.canlet.2019.09.006 |
| [14] |
D. Li, W. Chen, S.H. Liu, X. Chen, J. Yin, Chin. Chem. Lett. 31 (2020) 2891-2896. DOI:10.1016/j.cclet.2020.02.047 |
| [15] |
Q. Guo, Y. Zhang, Z.H. Lin, Q.Y. Cao, Y. Chen, Dyes Pigm. 172 (2020) 107872. DOI:10.1016/j.dyepig.2019.107872 |
| [16] |
K. Wang, D. Xi, C. Liu, et al., Chin. Chem. Lett. 31 (2020) 2955-2959. DOI:10.1016/j.cclet.2020.03.064 |
| [17] |
P. Lu, X. Zhang, T. Ren, L. Yuan, Chin. Chem. Lett. 31 (2020) 2980-2984. DOI:10.1016/j.cclet.2020.08.016 |
| [18] |
X. Li, H. Wang, Y. Zhang, Q. Cao, Y. Chen, Chin. Chem. Lett. 32 (2021) 1541-1544. DOI:10.1016/j.cclet.2020.10.047 |
| [19] |
F. Hou, L. Zhu, H. Zhang, et al., New J. Chem. 44 (2020) 1537-1541. DOI:10.1039/C9NJ05386G |
| [20] |
Y.L. Pak, J. Li, K.C. Ko, et al., Anal. Chem. 88 (2016) 5476-5481. DOI:10.1021/acs.analchem.6b00956 |
| [21] |
K. Zhong, S. Zhou, X. Yan, et al., Dyes Pigm. 174 (2020) 108049. DOI:10.1016/j.dyepig.2019.108049 |
| [22] |
K. Zhang, J. Zhang, Z. Xi, et al., Chem. Sci. 8 (2017) 2776-2781. DOI:10.1039/C6SC05646F |
| [23] |
W. Wang, C. Wu, C. Yang, et al., Sens. Actuators B: Chem. 255 (2018) 1953-1959. DOI:10.1016/j.snb.2017.08.222 |
| [24] |
S.J. Li, Y.F. Li, H.W. Liu, et al., Anal. Chem. 90 (2018) 9418-9425. DOI:10.1021/acs.analchem.8b02068 |
| [25] |
Q. Li, Z. Wang, M. Zhao, et al., Sens. Actuators B: Chem. 298 (2019) 126898. DOI:10.1016/j.snb.2019.126898 |
| [26] |
S. Muthusamy, K. Rajalakshmi, D. Zhu, et al., Sens. Actuators B: Chem. 320 (2020) 128433. DOI:10.1016/j.snb.2020.128433 |
| [27] |
H. Wang, D. Yang, R. Tan, et al., Sens. Actuators B: Chem. 247 (2017) 883-888. DOI:10.1016/j.snb.2017.03.030 |
| [28] |
C. Yan, L. Shi, Z. Guo, W. Zhu, Chin. Chem. Lett. 30 (2019) 1849-1855. DOI:10.1016/j.cclet.2019.08.038 |
| [29] |
M.H. Lee, J.L. Sessler, J.S. Kim, Acc. Chem. Res. 48 (2015) 2935-2946. |
| [30] |
Y.H. Zhang, X. Li, L. Huang, et al., Chem. Commun. 56 (2020) 10317-10320. DOI:10.1039/D0CC02045A |
| [31] |
W. Feng, C. Gao, W. Liu, et al., Chem. Commun. 52 (2016) 9434-9437. DOI:10.1039/C6CC02932A |
| [32] |
J.H. Wang, A.N. Endsley, C.E. Green, A.C. Matin, BMC Cancer 16 (2016)) 524. DOI:10.1186/s12885-016-2508-6 |
| [33] |
J. Zheng, Y. Liu, F. Song, et al., Chem. Commun. 56 (2020) 5819-5822. DOI:10.1039/D0CC02019B |
| [34] |
X. Lou, Z. Zhao, B.Z. Tang, Small 12 (2016) 6430-6450. |
| [35] |
D. Chen, W. Qin, H. Fang, et al., Chin. Chem. Lett. 30 (2019) 1738-1744. DOI:10.1016/j.cclet.2019.08.001 |
| [36] |
C. Jiang, L. Li, J. Jiang, et al., Chin. Chem. Lett. 31 (2020) 447-450. |
| [37] |
X.J. Zou, Y.C. Ma, L.E. Guo, et al., Chem. Commun. 50 (2014) 13833-13836. |