 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China
Hypericum (Hypericaceae) is a genus of flowering plants that comprises almost 500 species distributed in different temperate climatic regions all over the world [1]. These species include Hypericum beanii N. Robson (H. beanii), a perennial plant that originally grew in East Asia. The plants were used in traditional Chinese medicine for the treatment of wounds, abscesses, bleeding, cephalalgia, and abnormal menstruation [2, 3]. The compounds isolated from Hypericum plants include prenylated acylphloroglucinols [4-6], aromatic polyketides [7] (e.g., napthodianthrones, flavonoids, and xanthones), triterpenoids [8], meroterpenoids [9], procyanidins [10], dibenzofuran and pyranone [11]. However, it was reported that the components of H. beanii merely resolved some xanthones, triterpenoids, spirocyclic acylphloroglucinols, and monoterpenoid polyprenylated acylphloroglucinols [13-16]. In our current research, chemical investigation of H. beanii aerial parts have led us to the discovery of two new pairs of derivatives, with an unprecedented 6/6/4/6/6 polycyclic skeleton, namely (±)-hyperterpenoid A [(±)-1] and (±)-hyperterpenoid B [(±)-2] (Fig. 1). The strained cyclopropane and/or cyclobutane subunits occurred in many complex natural products, and these compounds exhibited versatile biological activities [12]. It was noteworthy that compoud (±)-1 and (±)-2 also obtain this cyclobutane motif. Their structures and absolute configurations were elucidated by extensive spectroscopic methods, electronic circular dichroism (ECD) calculations, and single-crystal X-ray diffraction experiments. Moreover, the related plausible biogenetic pathways were also presented. As for the biological activities, compound (±)-2, particularly (-)-2, demonstrated potential anti-inflammatory bioactivity, and other compounds, apart from (-)-2, were found to exhibit greater neuroprotective effects. Interestingly, the huge differences between the anti-inflammatory bioactivity results of the enantiomers and diastereomers may enlighten us in stucture-activity relationship to some extent.

|
Download:
|
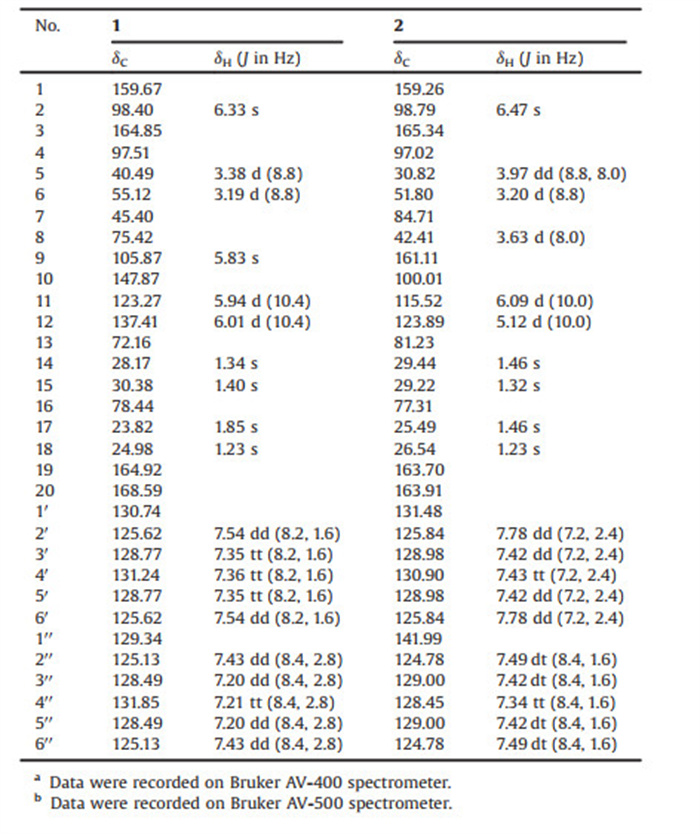
| Fig. 1. Structures of compounds 1-3. | |
Hyperterpenoid A was separated as colorless crystals (mp 122–124 ℃) that produced [M+H]+ ion in HRESIMS. The molecular formula of C32H28O6 as deduced from HRESIMS ion at m/z 509.1956 [M+H]+ (calcd. for C32H29O6, 509.1970), indicating 19 degrees of unsaturation. The IR spectrum displayed the characteristic absorption bands for carbonyl group (1725−1750 cm−1), benzene ring (1639 cm−1, 3000−3100 cm-1). The 1H NMR spectrum exhibited signals corresponding to four types of methyl protons [δH 1.23 (3H, s), 1.34 (3H, s), 1.40 (3H, s), 1.85 (3H, s)], four different environments of olefinic protons [δH 5.38 (1H, s), 5.94 (1H, d, J=10.4 Hz), 6.01 (1H, d, J=10.4 Hz), 6.33 (1H, s)], and two kinds of protons [δH 3.19 (1H, d, J=8.8 Hz), 3.38 (1H, d, J=8.8 Hz)] linked to oxygenated tertiary carbons and two monosubstituted benzene rings. The DEPT and HSQC spectra revealed 28 carbon resonances, corresponding to four sp3 primary carbons, three sp3 tertiary carbons, three oxygenated sp3 quaternary carbons, eight sp2 olefinic carbons and two ester carbonyl carbons (Table 1).
|
|
Table 1 1H (400MHz)a and 13C (125MHz)b NMR data of 1 and 2 in CDCl3. |
{kind=link}
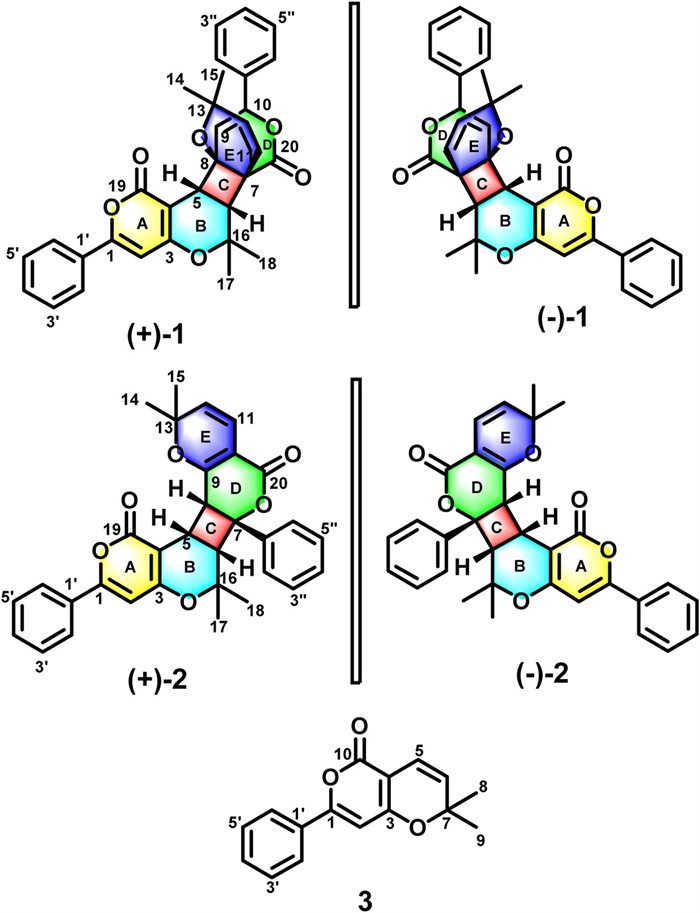
As shown in Fig. 2, the HMBC of H-2 to C-3, and C-4; H-5 to C-4, C-6 and C-19; H-6 to C-4, C-5 as well as the 1H-1H COSY cross-peaks of H-5/H-6 confirmed the formation of A/B rings. The 1H-1H COSY results also showed that H-5 and H-6 had identical coupling constants (resulting in the splitting of proton peaks into "d" peaks in 1H NMR spectroscopy), which indicated that C-5 and C-6 were adjacent carbon atoms surrounded by quaternary carbons. The HMBC correlations of H-5 to C-7, C-8, and C-9; H-6 to C-7, C-8, and C-20; and H-9 to C-7, and C-8 demonstrated that the C/D rings constituted a system that was fused at C-4 and C-6. The HMBC correlation experiments of H-11 to C-6, C-7, C-8, and C-13 suggested that C-11 was attached to a tertiary carbon atom (C-7). Therefore, the E ring of 1 was formed by joining the oxygenated tertiary C-8 (δC 75.4) and C-13 (δC 72.2) carbon atoms via an ether linkage.

|
Download:
|
| Fig. 2. Key 2D NMR correlations of 1. | |
{kind=link}
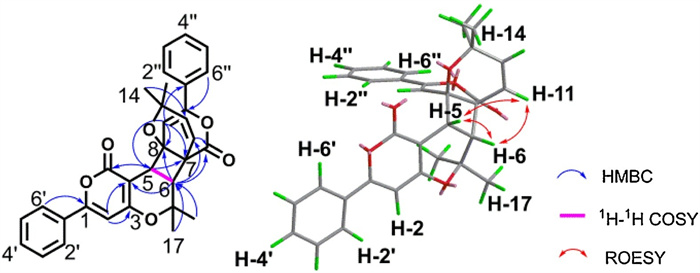
Providentially, single-crystal X-ray diffraction allowed for the explicit determination of the absolute configuration of (±)-1 (Fig. 3).

|
Download:
|
| Fig. 3. ORTEP(oak ridge thermal ellipsoid plot program) of the X-ray crystal structure of (±)-1 (ellipsoids are given at the 50% probability level). | |
{kind=link}
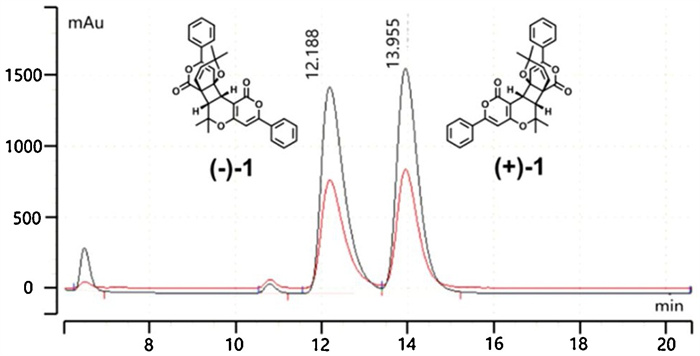
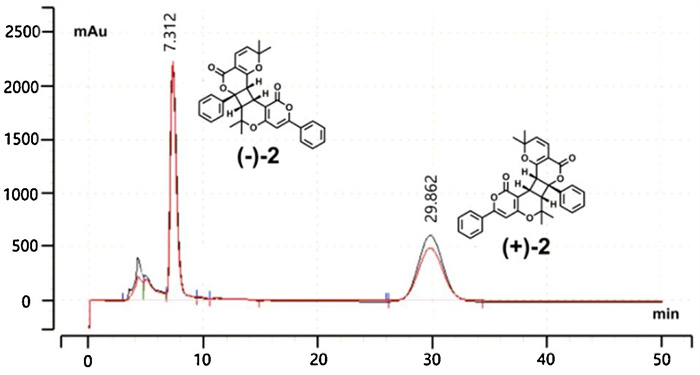
The racemate (±)-1 and (±)-2 were successfully separated into the two optically pure enantiomers (ee ≥ 99%) using a preparative HPLC system (flow rate=3.0 mL/min) that was equipped with a CHIRALPAK AD-H column (250 mm×10 mm, 5 μm) (Figs. 4 and 5).

|
Download:
|
| Fig. 4. HPLC separation chromatogram of 1 on chiral AD-H column (250 mm×10 mm, 5 μm). | |
{kind=link}

|
Download:
|
| Fig. 5. HPLC separation chromatogram of 2 on chiral AD-H column (250 mm×10 mm, 5 μm). | |
{kind=link}
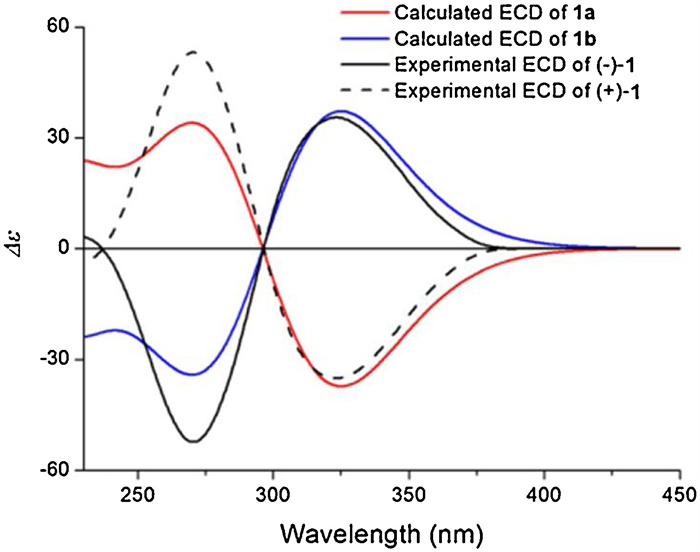
In order to confirm the absolue configuration, experimentally recorded ECD spectra were compared with those calculated theoretically to identify the absolute configurations of (+)-1 and (-)-1. The enantiomer pair proposed as model compounds were [(5R, 6R, 7R, 8S)-1 (1a) and (5S, 6S, 7S, 8R)-1 (1b)](Fig. S50 in Supporting information). By matching the experimentally recorded ECD spectra of the enantiomers to those predicted theoretically for the model compounds, it was determined that the absolute configurations of (+)-1 and (-)-1 are (5R, 6R, 7R, 8S)-1 (1a) and (5S, 6S, 7S, 8R)-1 (1b), respectively (Fig. 6).

|
Download:
|
| Fig. 6. Experimental ECD spectra of (+)-1 and (-)-1 and theoretical ECD spectra of 1a and 1b. | |
{kind=link}
Hyperterpenoid B [(±)-2] was isolated as a colorless oil. The molecular formula was C32H28O6 (m/z 509.1954 for the [M+H]+ ion C32H29O6, calcd. for 509.1970) based on HRESIMS analysis, which was identical to the formula of 1. The IR spectrum of (±)-2 showed absorption peaks corresponding to ester carbonyl group (1707 cm−1) and the benzene ring (1639 cm−1, 3000−3100 cm-1). The 1H NMR spectrum of (±)-2 (Table 1) indicated the presence of four types of methyl protons [δH 1.23 (3H, s), 1.32 (3H, s), 1.46 (3H, s), 1.46 (3H, s)], three types of olefinic protons [δH 5.12 (1H, d, J=10.0 Hz), 6.09 (1H, d, J =10.0 Hz), 6.47 (1H, s)], and three types of protons [δH 3.20 (1H, d, J=8.8 Hz), 3.63 (1H, d, J=8.0 Hz), 3.97 (1H, dd, J=8.8 Hz, 8.0 Hz)] bonded to tertiary carbons and two monosubstituted benzene rings. Combined with the DEPT and HSQC spectra, the 13C NMR spectroscopic results suggested the existence of three methines, four methyls, eight olefinic, twelve aromatic, and five quaternary (two ester carbonyl groups and three oxygenated quaternary carbons) carbon atoms. Furthermore, the chemical shift values of δC 163.7, 97.0, 165.3 and δC 163.9, 100.0, 161.1 indicated the presence of two α, β-unsaturated lactone fragments.
In addition to the 1H-1H COSY cross-peaks of H-5/H-6, the HMBC spectral correlations (Fig. 7) of H-2′ to C-1; H-2 to C-1′, C-1, C-3, and C-4; H-5 to C-4, C-6, C-16, and C-19; H-6 to C-4, C-5, C-17, and C-18 supported the formation of the A/B rings. Meanwhile, the 1H-1H COSY correlations of H-5/H-6/H-8 and the coupling constants of H-5 (dd, J=8.8 Hz, 8.0 Hz)/H-6 (d, J=8.8 Hz) and H-5(dd, J=8.8 Hz, 8.0 Hz) /H-8 (d, J=8.0 Hz) indicated that C-5 was adjacent to the C-6 and C-8 atoms that were surrounded by quaternary carbon atoms. Furthermore, the HMBC spectral correlations of H-5 to C-7, C-8, and C-9; H-6 to C-7, C-8, and C-1′′; H-8 to C-7, C-9, and C-10 demonstrated that the C/D rings constituted a 4/6 fused ring system similar to that observed in (±)-1. These correlations also confirmed the presence of a monosubstituted benzene ring attached to C-7. As for the correlations of H-12 to C-10, C-14, and C-15; H-11 to C-9 and C-20, they were consistent with the 1H-1H COSY cross-peaks of H-11/H-12 showing that the E and D rings were fused to each other, and that they shared the C-9 and C-10 atoms. The planar molecular structure of (±)-2 was thereby established.

|
Download:
|
| Fig. 7. Key 2D NMR correlations of 2. | |
{kind=link}
The relative configuration of compound (±)-2 [(±)-hyperterpenoid B] was determined by analyzing its ROESY spectrum. The correlations of H-5/H-6, H-5/H-8 and H-6, and H-8/H-2′′ and H-6′′ suggested that the hydrogen atoms H-5, H-6, and H-8, as well as the C7-C1′′ fragment are β-oriented. The ECD spectra indicated that (±)-2 did not exhibit a Cotton effect, and thus, it was a racemate. Chiral separation was achieved on a CHIRALPAK AD-H HPLC column, and the experimentally recorded ECD spectra of the enantiomers were compared to those calculated theoretically for the proposed model compounds [(5S, 6S, 7R, 8R)-2 (2a) and (5R, 6R, 7S, 8S)-2 (2b), Fig. S51 in Supporting information]. The results indicated that the absolute configurations of (+)-2 and (-)-2 are (5S, 6S, 7R, 8R)-2 (2a) and (5R, 6R, 7S, 8S)-2 (2b), respectively (Fig. 8).

|
Download:
|
| Fig. 8. Experimental ECD spectra of (+)-2 and (-)-2 and theoretical ECD spectra of 2a and 2b. | |
{kind=link}
The 1D and 2D NMR spectra of compound 3 were found to be identical to those reported previously for hypermonone A [17, 18]. However, this was the first time that compound 3 has been isolated from H. beanii.
The feasible biogenetic pathways of compounds 1-3 were presented in Scheme S1 (Supporting information). Biogenetically, the scaffold of this compound was similar to that of PPAPs (polycyclic polyprenylated acylphloroglucinols) [19], with the core moiety originating from typical polyketide-type biosynthetic processes comprising the condensation of one acyl-CoA and two malonyl-CoA units (as opposed to acylphloroglucinol across three malonyl-CoA units) [20, 21]. This core moiety undergoes prenylation, oxidation, cyclization, and dehydration reactions to form hypermonone A (3). The dimers of compound 3 [hyperterpenoid A (1) and hyperterpenoid B (2)] were biogenetically produced via the [2+2] cycloaddition of two molecules of this compound.
In preparation for HPLC-HRESIMS analysis, the aerial parts of the H. beanii plant were air-dried, powdered, extracted using MeOH at room temperature, and protected from light, according to the method described in previously published reports [22]. The ion peaks observed in the EIC (extracted-ion chromatogram) and LC–MS/MS spectra of compounds 1 and 2 demonstrated that these species were not isolation artifacts (Figs. S48 and S49 in Supporting information).
To assess the neuroprotective effects of compounds 1-3, they were modeled against glutamic acid and oxygen-glucose deprivation [20]. The results indicated that at a density of 10 μmol/L, the investigated compounds, (±)-1, (+)-1, (-)-1, (±)-2, (+)-2, (-)-2, and 3, against glutamic acid-induced toxicity in SK-N-SH cells, which improving cell viability from 54.3% to 61.0%, 60.7%, 61.8%, 60.7%, 57.7%, 57.8%, and 58.5%, respectively (Fig. S52 in Supporting information). They were also tested the ability to against (OGD)-induced in SK-N-SH cells, which improving cell viability from 52.9% to 53.0%, 50.6%, 53.5%, 63.6%, 47.7%, and 69.4%, respectively. Apart from (-)-2, other compounds were found to exhibit greater neuroprotective effects than the positive control drug, potassium 2-(1-hydroxypentyl)-benzoate (dl-PHPB) (60.7% and 57.3%).
Moreover, all of the isolates were evaluated for their inhibitory activity against the production of nitric oxide (NO) production in the polymorphonuclear leukocytes (induced using lipopolysaccharides) of mice. In fact, the administration of 10 μmol/L (±)-1, (+)-1, (-)-1, (±)-2, (+)-2, (-)-2, 3, and the positive control drug, dexamethasone (IC50 value: 0.326 μmol/L), inhibits NO production by 24.51%, 22.53%, 23.68%, 72.92%, 0.69%, 80.12% or 55.10%, respectively (Fig. S53 in Supporting information). Among them, compound (-)-2, demonstrated potential anti-inflammatory bioactivity, with an IC50 value of 3.14 μmol/L. This highlighted the significance of the compound (-)-2 as a template for the investigation of anti-inflammatory agents. Interestingly, the results also showed that not only were there huge differences between the two classes of compounds that were connected in different ways, but there were also significant differences between pairs of enantiomers that were connected in the same way. It should be noted that (+)-2 and (-)-2 presented significantly different anti-inflammatory properties, which necessitates further research regarding the binding of drugs to target proteins.
In conclusion, (±)-hyperterpenoids A and B (1 and 2), two pairs of polymeric pyrone meroterpenoid derivatives with an unprecedented 6/6/4/6/6 polycyclic skeleton, were isolated and identified from the extracts of the aerial parts of H. beanni for the first time. As is known to all, the natural polymeric compounds identified to date are numerous [16] and these compounds are characterized by varying bonding regimes. Many of them were associated with [2+2] or [4+2] cycloaddition compounds polymerized from achiral monomers [22]. In this study, we also revealed how hyperterpenoid A (1) and hyperterpenoid B (2) were biogenetically produced via the [2+2] cycloaddition of two molecules of compound 3. Interestingly, (+)-2 and (-)-2 present different anti-inflammatory properties. The potential anti-inflammatory function of (-)-hyperterpenoid B may lead us to new discovery of structure activity relationship between racemates, enantiomers, and diastereomers, as well as provides a template for further study in the field of medicinal chemistry.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe project was financially supported by Projects of International Cooperation and Exchanges NSFC (NSFC-VR, No. 81361138020), National Science and Technology Major Projects for "Major New Drugs Innovation and Development", Research and Development of New Drug Varieties from Natural Product Sources and Their Key Innovative Technological Systems (Nos. 2018ZX09711001-001-001 and 2018ZX09711001-001-003) and the CAMS Innovation Fund for Medical Sciences (CIFMS), the CAMS Initiative for Innovative Medicine (CAMS-I2M, No. 2016-I2M-1-010). Wen-Yi He, Ming-Bao Lin, Wei-Ping Wang and Jian-Bei Li are responsible for collecting the NMR spectrum, responsible for the biological assay, and support for providing the HPLC analytical method.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.027.
| [1] |
N.M. Nürk, S. Madriñán, M.A. Carine, M.W. Chase, F.R. Blattner, Mol. Phylogenet. Evol. 66 (2013) 1-16. DOI:10.1016/j.ympev.2012.08.022 |
| [2] |
W.K. Shiu, S. Gibbons, Phytochemistry 67 (2006) 2568-2572. DOI:10.1016/j.phytochem.2006.09.037 |
| [3] |
G.P. Zhao, S. Dai, R.S. Chen, Dictionary of Traditional Chinese Medicine, Vol. 1. Shanghai Science and Technology Publishing House, Shanghai, 2006. pp. 1396-1397.
|
| [4] |
W. Gao, J.W. Hu, W.Z. Hou, et al., Tetrahedron Lett. 57 (2016) 2244-2248. DOI:10.1016/j.tetlet.2016.04.026 |
| [5] |
W. Gao, W.Z. Hou, J. Zhao, et al., J. Nat. Prod. 79 (2016) 1538-1547. DOI:10.1021/acs.jnatprod.5b01063 |
| [6] |
J.W. Hu, M.J. Shi, J.J. Wang, et al., J. Nat. Prod. 81 (2018) 2348-2356. DOI:10.1021/acs.jnatprod.8b00176 |
| [7] |
Pinarosa Avato, Stud. Natl. Prod. Chem. 30 (2005) 603-634. DOI:10.1016/s1572-5995(05)80043-2 |
| [8] |
C. Chen, G. Wei, H. Zhu, Y. Guo, et al., Fitoterapia 103 (2015) 227-230. DOI:10.1016/j.fitote.2015.04.009 |
| [9] |
N. Tanaka, M. Okasaka, Y. Ishimaru, et al., Org. Lett. 7 (2005) 2997-2999. DOI:10.1021/ol050960w |
| [10] |
O. Ploss, F. Petereit, A. Nahrstedt, Pharmazie 56 (2001) 509-511. DOI:10.1023/A:1011222719485 |
| [11] |
W.K.P. Shiu, Simon Gibbons, Phytochemistry 70 (2009) 403-406. DOI:10.1016/j.phytochem.2008.12.016 |
| [12] |
Y.Y. Fan, X.H. Gao, J.M. Yue, Sci. China Chem. 59 (2016) 1126-1141. DOI:10.1007/s11426-016-0233-1 |
| [13] |
W.K.P. Shiu, S. Gibbons, Phytochemistry 67 (2006) 2568-2572. DOI:10.1016/j.phytochem.2006.09.037 |
| [14] |
X.Q. Chen, Y. Li, K.Z. Li, et al., Chem. Pharm. Bull. 59 (2011) 1250-1253. DOI:10.1248/cpb.59.1250 |
| [15] |
D.S. Yang, Z.L. Li, Y.P. Yang, X.L. Li, W.L. Xiao, Chin. Herb. Med. 7 (2015) 375-379. DOI:10.1016/S1674-6384(15)60067-3 |
| [16] |
Y.R. Li, W.J. Xu, S.S. Wei, et al., Phytochemistry 159 (2019) 56-64. DOI:10.1109/iccd46524.2019.00016 |
| [17] |
H.C. Shen, J. Wang, K.P. Cole, et al., J. Org. Chem. 68 (2013) 1729-1735. |
| [18] |
Y.R. Zeng, L.P. Wang, Z.X. Hu, et al., Fitoterapia 125 (2018) 59-64. DOI:10.1016/j.fitote.2017.12.013 |
| [19] |
X.W. Yang, R.B. Grossman, G. Xu, Chem. Rev. 118 (2018) 3508-3558. DOI:10.1021/acs.chemrev.7b00551 |
| [20] |
Y.T. Zhang, F.M. Li, Y.Z. Guo, et al., Eur. J. Pharmacol. 792 (2016) 48-53. DOI:10.1016/j.ejphar.2016.10.029 |
| [21] |
Y.L. Zhang, X.W. Zhou, X.B. Wang, et al., Org. Lett. 19 (2017) 3013-3016. DOI:10.1021/acs.orglett.7b01276 |
| [22] |
J. Zou, G.D. Chen, H. Zhao, et al., Org. Lett. 20 (2018) 884-887. DOI:10.1021/acs.orglett.8b00017 |