 2021, Vol. 32
2021, Vol. 32 


b College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
c School of Environment and Civil Engineering, Dongguan University of Technology, Dongguan 523808, China
Lithium-ion batteries (LIBs) have been widely applied in portable devices and electric vehicles due to their high power density, high energy density, low self-discharge, and portability [1-5]. Particularly, LiCoO2 material is extensively employed for LIBs because of its high voltage, high density, and excellent electrochemical performance [6, 7]. However, the life span of LIBs is generally 4–6 years [8]. Spent LiCoO2 batteries will not only waste of Li and Co but also cause serious environmental problems if they are not handled properly [9-12]. Therefore, it is of great significance to recover valuable metals from spent LiCoO2 batteries in terms of both resource conservation and sustainable economic development.
Metallurgical technological means involving pyrometallurgy and hydrometallurgy play a significant role in the field of recycling spent LIBs [13]. In particular, hydrometallurgy is more suitable for recycling spent batteries because of its low energy consumption and no loss of Li volatilization due to high temperature [14, 15]. Hydrometallurgy dissolves valuable metals into lixivium, followed by recovering them [16]. Acid leaching of LiCoO2 material becomes a research hotspot for its high efficiency and rapid reaction [17]. Compared with inorganic acid leaching (such as H2SO4 [18], HNO3 [19] and H3PO4 [20]), organic acid leaching (such as acetic acid [21] and ascorbic acid [22]) is widely used in recover valuable metals because it did not produce excessive acid wastewater and corrosion equipment. Correspondingly, there are other elements such as Al, Fe and Cu in the leaching solution in addition to Co and Li due to the poor selectivity of the acid leaching process [23, 24]. So this will inevitably increase the difficulty of subsequent purification and separation process [25]. Recently, ammonia leaching has undergone a flurry of research owing to its good selectivity and retrievability [26-31]. Chao Wang et al. proposed an NH3-(NH4)2CO3-Na2SO3 leaching system to recover cathode material from commercial LiNixCoyMn1-x-yO2 (NCM) and spent NCM. 79.1% of the lithium, 86.4% of the cobalt, and 85.3% of the nickel were selectively leached under the optimum condition, respectively [32]. Yaping Qi et al. used ammonia leaching method to recover Co and Li from spent LIBs. This method yields a Co and Li leaching efficiency of 91.16% and 97.57%, respectively [33].
Ammonia has a strong selectivity for the valuable metals, such as Ni and Co, to form the metal complexes (Co(NH3)n2+/Ni(NH3)n2+) in the leaching solution according to the Eh-pH diagram for Co-NH3-H2O and Ni-NH3-H2O system, so that the undesirable impurities, such as Al, Fe and Mn, are hardly leached as precipitates to separate [34]. At present, most ammonia leaching research focused on NCM/LiNixCoyAl1-x-yO2 (NCA), but few on LiCoO2 material. Ammonia leaching of LiCoO2 material is more difficult than NCM/NCA, mainly because LiCoO2 is a primary particle and NCM/NCA is a secondary particle composed of primary grains. When NCM is leached by an ammonia leaching agent, the secondary particles will break, resulting in an increase in the specific surface of the reaction, which is more conducive to the ammonia leaching. As the complexity of the cathode electrode composition of LIBs (LiCoO2/NCM/NCA/LiMn2O4 mixture) increases, it is necessary to overcome the issue of ammonia leaching of LiCoO2 if this method is used to recover waste LIBs in the future. Therefore, it will improve the applicability of the ammonia leaching method and promote the development of the new energy industry chain if this method can be applied to LiCoO2 material. In addition, the ammonia leaching method used to recover spent LiCoO2 batteries has inherent advantages. Compared with NCM/NCA, ammonia leaching LiCoO2 material will not generate Mn/Al-containing solid slag, which is beneficial to the leaching process and does not require the slag treatment step. In this study, the leaching mechanism of LiCoO2 in ammonia-sodium sulfite-ammonium chloride was first elucidated. The leaching behavior of LiCoO2 material was revealed by studying the role of each component and the change of valence state of Co during the ammonia leaching process. According to the characteristics of small specific surface area and stable structure of LiCoO2, mechanical activation (high-energy ball milling) pretreatment was used to ameliorate its specific surface area and structure drawbacks, so as to improve the reaction rate. Finally, the apparent leaching kinetics models of Li and Co were established through the shrinking core reaction model and the apparent activation energies were also calculated. This research is of great significance for the efficient recovery of valuable elements from waste LiCoO2 batteries.
Spent LiCoO2 material was supplied by Huayou Cobalt Co., Ltd. The raw material was milled with 0.2 mm zirconia at 1650 r/min in aqueous solution for a certain amount of time (SPT 0.5 W, Shenzhen Samsung Feilong Machinery Co., Ltd.). All chemicals were of analytical grade. 3 g LiCoO2 powder was added to a 300 mL leaching agent heated to a certain temperature with a stirring speed of 400 rpm. The leaching agent was composed of 4 mol/L ammonia, 0.5 mol/L sodium sulfite, and 1.5 mol/L ammonium chloride. The leaching solution was separated from the leaching residue after a certain time. Then, the concentration of Li and Co in the solution was analyzed by ICP-OES, and the leaching efficiency was calculated [21]. The leaching residues were dried at 80 ℃ for 24 h and then analyzed. In order to explore the mechanism of LiCoO2 ammonia leaching, the following different kinds of leaching agent (with the same concentration as above) were used for the tests: 1) H2O, 2) NH3·H2O, 3) Na2SO3, 4) NH3·H2O, 5) NH3·H2O+Na2SO3 + NH4Cl. It should be noted that the raw leaching powder of experiment 4 was used from the residue of experiment 3, and the other LiCoO2 powder was milled for 5 h.
The crystal structure of powders and leaching residues were characterized by X-ray diffraction (XRD: TTR-Ⅲ, Japan) measurement with Cu-Kα in the range 10° ≤ 2θ ≤ 80°. Morphologies and sizes of the particles were observed by field-emission scanning electron microscope (FESEM: TESCAN MIRA3 LMU, Czech) and transmission electron microscope (TEM: Tecnai G2 F20 S-TWIN, Japan). The particle size distribution data measured on a laser diffraction particle size analyzer (Bettersize2000N, China). The analysis of metal ions was used by plasma-atomic emission spectrometry (ICP: ICAP7400 Radial, America). The residues oxidation states were detected by X-ray photoelectron spectroscopy (XPS: Escalab 250xi, America).
Li and Co were barely leaching out when deionized water, ammonia, and sodium sulfite were used as single leaching reagent (Fig. 1a). The mechanism of ammonia leaching LiCoO2 was studied by detecting the change of Co valence state (Figs. 1b and e). It can be seen from Fig. 1b-1 and 2 that Co on the surface of LiCoO2 particles is basically +3 and does not react with ammonia. Sodium sulfite can reduce Co3+ on the surface of LiCoO2 particles to Co2+ (Fig. 1b-3), and then the reduced Co2+ can dissolve in ammonia (Fig. 1b-4). It indicates that sodium sulfite reduces Co3+ on the surface of LiCoO2 particles to Co2+, and then Co2+ leached by ammonia. To sum up, the ammonia reduction leaching process of LiCoO2 repeats the above two steps until it is completely dissolved. The surface of leaching residue with sodium sulfite and ammonia has Co2+ and Co3+ (Fig. 1b-5). It demonstrates that ammonia dissolving Co2+ is slower than that of sodium sulfite reducing LiCoO2 particles.

|
Download:
|
| Fig. 1. (a) Leaching efficiency of Li and Co. (b) XPS spectra of residues leached under different leaching systems: (1) H2O, (2) NH3·H2O, (3) Na2SO3, (4) NH3·H2O, (5) NH3·H2O+Na2SO3. (c) Effect of leaching system on leaching efficiency of metals for 2 h, (d) pH at different systems: (1) Na2SO3 + NH3·H2O, (2) Na2SO3 + NH4Cl+NH3·H2O, (3) Na2SO3 + NH3·H2O regulated by H2SO4 and (e) schematic diagram of ammonia leaching LiCoO2 material. | |
{kind=link}
The leaching efficiencies of Li and Co were only 13.09% and 12.92% after 2 h when ammonia and sodium sulfite were used as leaching reagent (Fig. 1c-1), and the pH value of the solution was about 13 during the leaching process (Fig. 1d-1). Correspondingly, the leaching efficiencies of Li and Co increased to 77.01% and 89.27% after 2 h when ammonia, ammonium chloride (Fig. 1c-2), and sodium sulfite were used as leaching reagent, and the pH value of the solution was about 10 during the leaching process (Fig. 1d-2). To clarify the role of pH, the pH value of the leaching agent containing sodium sulfite and ammonia was adjusted to 10 by slowly adding sulfuric acid after 1 h from the start of the reaction (Fig. 1d-3). The leaching efficiency of Li and Co was significantly improved to 60.44% and 70.74%, respectively (Fig. 1c-3).
When ammonia, sodium sulfite, and ammonia chloride were used as leaching agents, the pH of the system was (Eq. 1) [35]:
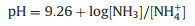
|
(1) |
The following reaction occurs under the system (Eq. 2):

|
(2) |
NH4+ and NH3 are a pair of conjugate acid-base. According to Eh-pH of Co-NH3-H2O [36], ammonium chloride makes the pH of the solution in the stability area of [Co(NH3)n]2+ and controls the stable concentration of ammonia in the leaching solution.
In conclusion, the ammonia leaching of LiCoO2 in NH3-Na2SO3-NH4Cl system is as follows (Eq. 3):
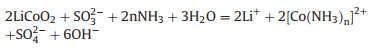
|
(3) |
In order to study the influence of high-energy ball milling on properties, a variety of methods were used for characterization. The absence of new peaks in high-energy ball milling indicates that no new phase is generated (Figs. 2a and a1). The intensity of the diffraction peak decreases and its width also widens with the increase of milling time, indicating that the crystallinity of the material decreased. The energy provided by the ball milling destroys the long-range ordered structure of the crystal. The crystallite size and microcosmic strain of LiCoO2 were calculated by Williamson-Hall method. After ball-milling for 5 h, the crystallite size and microcosmic strain changed from 2944 nm to 219 nm and 0.0093% to 0.2604%, respectively (Fig. 2b). The decrease of particle size will lead to an increase in the specific area of the reaction. The increase of microcosmic strain indicates the increase of defect concentration between grains and in the grain, which can reduce reaction energy barrier and facilitate LiCoO2 leaching [37].

|
Download:
|
| Fig. 2. (a) and (a1) XRD patterns of LiCoO2 at different ball milling time. (b) Variation of crystallite size (D) and microstrain (ε) of LiCoO2 with ball milling time. (c) Particle size distribution of LiCoO2 at different ball milling time. (d) TEM image of LiCoO2 ball milled for 5 h. (e) SEM images of LiCoO2 for high-energy ball milling time: (1-1), (1-2) untreated, (2) 0.5 h, (3) 1 h, (4) 2 h and (5) 5 h. | |
{kind=link}
As can be seen from Fig. 2e, the raw LiCoO2 particles are about 10−30 μm primary particles. After 5 h milling, the particles become sub-micron particles and agglomerate significantly. This is because the particles are too small and agglomerate can reduce the specific surface energy of the particles. The particle size distribution of LiCoO2 at different high-energy ball milling times indicated that ball milling could continuously reduce the particle size of LiCoO2 within 2 h, and the particle size remained stable at about 440 nm after ball milling for 5 h (Fig. 2c). LiCoO2 particles showed partial amorphous structure after ball-milling for 5 h (Fig. 2d). The amorphous structure has higher energy than its crystalline structure, which reduces the reaction energy barrier and facilitates the leaching process.
Fig. S1 (Supporting information) shows that the ammonia leaching efficiency of LiCoO2 material without milling is slow. The ammonia leaching efficiency of LiCoO2 increased with the increase of temperature, but the ammonia volatilization increased with the increase of temperature, and cobalt ammonia complex could not be stable at high temperatures, so the reaction temperature range was selected as 50–80 ℃. It took 48 h to reach the leaching equilibrium at 80 ℃, and the leaching efficiencies of Li and Co were 69.86% and 70.80%, respectively.
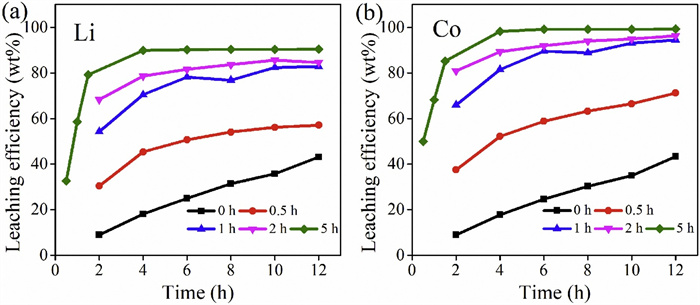
The leaching efficiencies continued to critically increase with the increase of ball milling time. The leaching speed is obviously accelerated with the increase of milling time. For LiCoO2 material ball milling for 5 h, the equilibrium is reached after leaching for 4 h and the leaching efficiency of Li and Co was 89.86% and 98.22%, respectively (Fig. 3). Compared with untreated LiCoO2 materials, the leaching time reduces drastically and the leaching efficiency of Li and Co is increased by 20.00% and 27.42%, respectively. It can be seen from the residue XRD and SEM (Fig. S2 in Supporting information) that residue has relatively high crystallinity and large particle is difficult to leach, resulting in that the LiCoO2 material cannot be completely leached and unleached Li and Co are in the residue (Fig. S3 in Supporting information).

|
Download:
|
| Fig. 3. Effect of ball milling time on leaching efficiency of LiCoO2 metals at different time intervals: (a) Li and (b) Co. | |
{kind=link}
To determine the ammonia leaching kinetics of untreated and ball milling for 5 h LiCoO2, the ammonia leaching data were fitted based on the surface chemical control (Eq. 4) and the diffusion control model (Eq. 5). The models are as follows:

|
(4) |
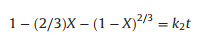
|
(5) |
where X is the fraction of the leaching metal; k1 and k2 are the apparent constant of different steps; and t is time.
The apparent activation energy of Li and Co is calculated according to the Arrhenius equation (Eq. 6):
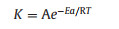
|
(6) |
where K is the reaction rate constant; A is the frequency factor; Ea is the apparent activation energy; R is the gas constant (8.314 J mol−1 K−1); T is Kelvin temperature.
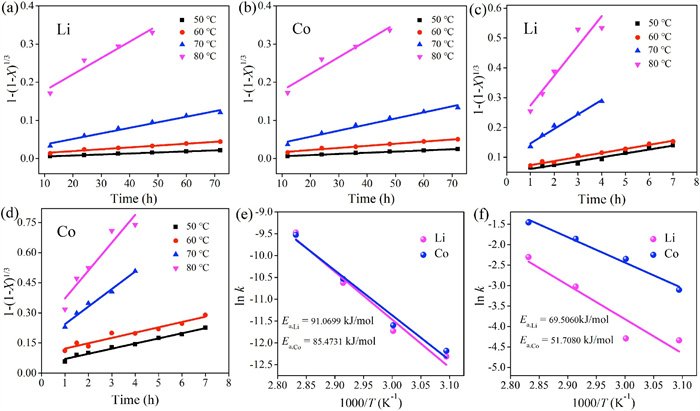
The ammonia leaching data in Figs. S1 and S4 (Supporting information) were fitted by the above models of the equation (Eqs. 4 and 5). The experimental leaching data of Li and Co match the chemical reaction control model (Fig. 4). The apparent activation energies of Li and Co leached from raw LiCoO2 were to be 91.1 kJ/mol and 85.5 kJ/mol, respectively. In contrast, the apparent activation energies of Li and Co leached from LiCoO2 after ball milling for 5 h is 69.5 kJ/mol and 51.7 kJ/mol, respectively. The ball milling process reduces the apparent activation energy of LiCoO2 in ammonia leaching.

|
Download:
|
| Fig. 4. (a) Li and (b) Co chemical control model for kinetics of leaching of untreated LiCoO2 metals. (c) Li and (d) Co chemical control model for kinetics of leaching of LiCoO2 metals ball milled for 5 h at different temperatures. (e) and (f) Arrhenius plot for the leaching of untreated LiCoO2 metals and LiCoO2 metals ball milled for 5 h in the temperature range 323.15–353.15 K. | |
{kind=link}
Apparent activation energy has a definite correlation with crystallinity and crystal form. The reduction of crystallinity and microstructure damage reduced the energy barrier required for the ammonia leaching of LiCoO2. So the leaching process is accelerated as the apparent activation energy decreases.
In summary, Li and Co were recovered from spent LiCoO2 batteries by combining ammonia leaching with high-energy ball milling. High-energy ball milling can refine the particles and reduce the crystallinity of LiCoO2, thus increasing the specific surface area of the reaction and reducing the reaction activation energy. Compared with raw LiCoO2 material, the leaching time of LiCoO2 after milling for 5 h was shortened from 48 h to 4 h, and the leaching efficiency of Li and Co increased from 69.86% and 70.80% to 89.86% and 98.22%, respectively. Finally, the apparent activation energy and the kinetics equation of leaching were obtained by the shrinkage nuclear reaction model, which showed that the reaction was controlled by chemical reaction. In the next study, we will focus on the ammonia leaching behavior of LiNixCoyMn1-x-yO2/LiNixCoyAl1-x-yO2 and LiCoO2 mixture.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 51822812, 51778627) and the Fundamental Research Funds for the Central Universities of Central South University (No. 2020zzts474).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2020.11.074.
| [1] |
J. Tao, G. Liu, Y. Chen, et al., RSC Adv. 8 (2018) 42438-42445. DOI:10.1039/c8ra08869a |
| [2] |
X. Ye, Z. Lin, S. Liang, et al., Nano Lett. 19 (2019) 1860-1866. DOI:10.1021/acs.nanolett.8b04944 |
| [3] |
C. Dun, G. Xi, T. Zhao, et al., Ceram. Int. 44 (2018) 9276-9282. DOI:10.1016/j.ceramint.2018.02.139 |
| [4] |
G. Zhang, Y. He, H. Wang, et al., J. Cleaner Prod. 231 (2019) 1418-1427. DOI:10.1016/j.jclepro.2019.04.279 |
| [5] |
X. Kong, J. Zhang, J. Huang, et al., Chin. Chem. Lett. 30 (2019) 771-774. DOI:10.1016/j.cclet.2018.10.006 |
| [6] |
J. Li, G. Wang, Z. Xu, J. Hazard. Mater. 302 (2016) 97-104. DOI:10.1016/j.jhazmat.2015.09.050 |
| [7] |
Z. Jian, W. Wang, M. Wang, et al., Chin. Chem. Lett. 29 (2018) 1768-1772. DOI:10.1016/j.cclet.2018.11.002 |
| [8] |
B. Wang, X.-Y. Lin, Y. Tang, et al., J. Power Sources 436 (2019) 226828. DOI:10.1016/j.jpowsour.2019.226828 |
| [9] |
G. Xi, L. Wang, T. Zhao, J. Magn. Magn. Mater. 424 (2017) 130-136. DOI:10.1016/j.jmmm.2016.10.031 |
| [10] |
W. Wang, Y. Zhang, X. Liu, S. Xu, ACS Sustainable Chem. Eng. 7 (2019) 12222-12230. |
| [11] |
G.P. Nayaka, K.V. Pai, J. Manjanna, S.J. Keny, Waste Manage. 51 (2016) 234-238. DOI:10.1016/j.wasman.2015.12.008 |
| [12] |
J.B. Dunn, L. Gaines, J. Sullivan, M.Q. Wang, Environ. Sci. Technol. 46 (2012) 12704-12710. DOI:10.1021/es302420z |
| [13] |
L. Zhuang, C. Sun, T. Zhou, et al., Waste Manage. 85 (2019) 175-185. DOI:10.1016/j.wasman.2018.12.034 |
| [14] |
X. Zhang, L. Li, E. Fan, et al., Chem. Soc. Rev. 47 (2018) 7239-7302. DOI:10.1039/c8cs00297e |
| [15] |
E. Fan, L. Li, Z. Wang, et al., Chem. Rev. 120 (2020) 7020-7063. DOI:10.1021/acs.chemrev.9b00535 |
| [16] |
J. Zhang, J. Hu, W. Zhang, et al., J. Cleaner Prod. 204 (2018) 437-446. DOI:10.1016/j.jclepro.2018.09.033 |
| [17] |
Y. Tang, H. Xie, B. Zhang, et al., Waste Manage. 97 (2019) 140-148. DOI:10.1016/j.wasman.2019.08.004 |
| [18] |
J. Hu, J. Zhang, H. Li, et al., J. Power Sources 351 (2017) 192-199. DOI:10.1016/j.jpowsour.2017.03.093 |
| [19] |
J. Guan, Y. Li, Y. Guo, et al., ACS Sustainable Chem. Eng. 5 (2017) 1026-1032. DOI:10.1021/acssuschemeng.6b02337 |
| [20] |
X. Chen, L. Cao, D. Kang, et al., Waste Manage. 80 (2018) 198-210. DOI:10.1016/j.wasman.2018.09.013 |
| [21] |
Y. Yang, X. Meng, H. Cao, et al., Green Chem. 20 (2018) 3121-3133. DOI:10.1039/C7GC03376A |
| [22] |
L.P. He, S.Y. Sun, Y.Y. Mu, et al.
, ACS Sustainable Chem. Eng. 5 (2017) 714-721. DOI:10.1021/acssuschemeng.6b02056 |
| [23] |
T. Zhang, Y. He, L. Ge, et al., J. Power Sources 240 (2013) 766-771. DOI:10.1016/j.jpowsour.2013.05.009 |
| [24] |
T. Zhang, Y. He, F. Wang, et al., Waste Manage. 34 (2014) 1051-1058. DOI:10.1007/978-3-642-54927-4_100 |
| [25] |
H. Chen, X. Zhu, Y. Chang, et al., Mater. Lett. 218 (2018) 40-43. DOI:10.3928/01484834-20180102-08 |
| [26] |
K. Meng, Y. Cao, B. Zhang, et al., ACS Sustainable Chem. Eng. 7 (2019) 7750-7759. DOI:10.1021/acssuschemeng.8b06675 |
| [27] |
H. Wang, K. Huang, Y. Zhang, et al., ACS Sustainable Chem. Eng. 5 (2017) 11489-11495. DOI:10.1021/acssuschemeng.7b02700 |
| [28] |
H. Ku, Y. Jung, M. Jo, et al., J. Hazard. Mater. 313 (2016) 138-146. DOI:10.1016/j.jhazmat.2016.03.062 |
| [29] |
X. Zheng, W. Gao, X. Zhang, et al., Waste Manage. 60 (2017) 680-688. DOI:10.1016/j.wasman.2016.12.007 |
| [30] |
C. Wu, B. Li, C. Yuan, et al., Waste Manage. 93 (2019) 153-161. DOI:10.1016/j.wasman.2019.04.039 |
| [31] |
Q. Li, K.Y. Fung, K.M. Ng, ACS Sustainable Chem. Eng. 7 (2019) 12718-12725. DOI:10.1021/acssuschemeng.9b00590 |
| [32] |
C. Wang, S. Wang, F. Yan, et al., Waste Manag. 114 (2020) 253-262. DOI:10.1016/j.wasman.2020.07.008 |
| [33] |
Y. Qi, F. Meng, X. Yi, et al., J. Cleaner Prod. 251 (2019) 119665. |
| [34] |
Y. Ma, J. Tang, R. Wanaldi, et al., J. Hazard. Mater. 402 (2020) 123491. DOI:10.1016/j.jhazmat.2020.123491 |
| [35] |
R. Das, S. Anand, S. Das, P. Jena, Hydrometallurgy 16 (1986) 335-344. DOI:10.1016/0304-386X(86)90008-3 |
| [36] |
X. Meng, K.N. Han, Miner. Process. Extr. Metall. Rev. 16 (1996) 23-61. DOI:10.1080/08827509608914128 |
| [37] |
Y. Tiechui, C. Qinyuan, L. Jie, Hydrometallurgy 104 (2010) 136-141. DOI:10.1016/j.hydromet.2010.05.008 |