 2021, Vol. 32
2021, Vol. 32 


b Pharmaceutical and Medicinal Chemistry, Saarland University, Campus C2.3, D-66123, Saarbrücken, Germany
Beyond its classical physiological functions in regulating homeostasis of water and electrolytes and thus modulating blood volume, aldosterone was identified recently as a deleterious factor when exorbitant [1]. This principle mineralocorticoid potently induces reactive oxygen species and inflammation stimulating vascular stiffening and vasoconstriction. Moreover, aldosterone up-regulates a series of genes promoting fibrosis, which, accompanied by inflammation as an accomplice, cause fibroblast proliferation, myocyte necrosis and collagen synthesis, resulting in cardiac fibrosis. These pathological alterations trigger cardiac hypertrophy and ventricular remodeling, consequently leading to congestive heart failure [2]. Besides, Aldosterone augments the expression of various prosclerotic growth factors, such as transforming growth factor β1 and plasminogen activator inhibitor 1, which, together with fibrotic effects and chronic inflammation, inflict tubular damage, glomerular injury and renal fibrosis [3].
As the effects of aldosterone are mainly mediated by mineralocorticoid receptor (MR), its antagonists like eplerenone and esaxerenone are employed in clinic for the treatment of hypertension, heart failure [4], chronic kidney disease and diabetic nephropathy [5]. However, these drugs lead to the accumulation of renin and aldosterone giving rise to resistance. More importantly, MR antagonism is impotent on the rapid nongenomic effects mediated by G protein-coupled estrogen receptor 1 (GRP30) [6] or interactions with leptin [7]. The inhibition of aldosterone synthase (CYP11B2), in contrast, could be a more promising treatment for these aldosterone related diseases. As CYP11B2 is the pivotal enzyme in aldosterone biosynthesis, its inhibition significantly reduces the plasma level of aldosterone. Although various CYP11B2 inhibitors have been identified recently [8], only few drug candidates proceed into clinical trials, but are discontinued later. This fact indicates the huge challenges in developing selective CYP11B2 inhibitors and, at the meantime, the urgent needs in designing chemical entities with novel structural scaffolds.
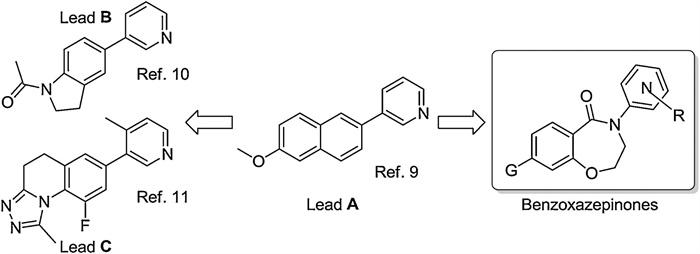
Our group has reported a series of pyridyl substituted naphthalenes as CYP11B2 inhibitors (Lead A, Scheme 1), [9] despite of excellent potency and selectivity, whose drug-likeness was, unfortunately, compromised due to poor pharmacokinetic properties. These drawbacks were caused by low water solubility and fast metabolism by hepatic CYP1A2, both of which were probably repercussions of the flat and conjugated naphthalene core. To solve this problem, saturation of the fused ring at the far-end to the pyridyl was attempted to interrupt the flat conformation leading to indolines [10] and dihydro-[1,2,4]triazolo[4,3-a]quinolones [11] (Leads B and C, respectively, in Scheme 1). Encouraged by the improvements observed, herein we further saturated the fused ring adjacent to pyridyl in the naphthalenen lead compound. After expanding this ring into a heptocycle and inserting an amido moiety, a series of pyridyl substituted 3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-ones were designed (Scheme 1). Compared to the rigid biaryl structures in lead compounds, the N-pyridyl cycloamido provided more flexibility to facilitate the molecule fitting into the active site of CYP11B2. This concept was subsequently demonstrated with a docking study of the design benzoxazepinones into CYP11B2 crystal (PDB ID: 4DVQ [12]). As exemplified with compound 5x, the twisted conformation of heptacycloamido guaranteed this compound to adopt a similar binding mode identified for indolines and dihydrotriazoloquinolones (Fig. 1), in which the pyridyl group coordinated to the heme iron in a nearly perpendicular manner with its sp2 hybrid N; the placement of pyridyl was further stabilized by a perpendicular π-π interaction with Phe130. The benzoxazepinone core, on the other hand, leaned to the β1-β3 sheets, similar to the natural substrate deoxycorticosterone, while π-π interactions between the benzoxazepinone core and Phe381 (perpendicular) and Phe487 (parallel) anchored binding and granted further affinity. Crucial H-bond was observed between the O in benzoxazepinone core and Tyr485. As the molecule pointed to Glu383 and Met111, various groups with the ability to form H-bond were probed in different proximity in order to promote affinity.

|
Download:
|
| Scheme 1. The design of pyridyl benzoxazepinones by saturating the fused ring adjacent to pyridyl in the naphthalene lead compound. | |

|
Download:
|
| Fig. 1. The binding of pyridyl substituted benzoxazepinones in CYP11B2 crystal (PDB ID: 4DVQ), illustrated with compound 5x (magenta): (A) top view; (B) side view, with a close vision of potential H-bonds between the piperazinyl group and Glu383 and Met111, as well as a salt bridge to Glu383 (distances around 3 and 2.8 Å, respectively). | |
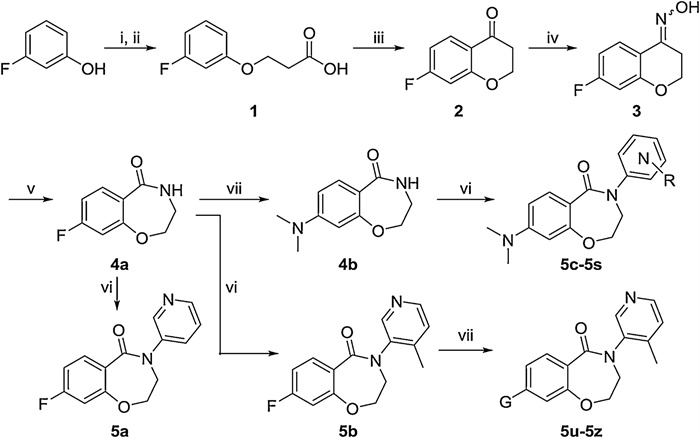
The designed compounds were substantiated via a concise synthetic route (Scheme 2). The starting material 3-fluorophenol was coupled with methyl 3-bromopropanoate, which was subsequently fused to the phenyl part forming a chroman-4-one scaffold (2) in an acidic environment. After it was converted into oxime intermediate 3, a Beckmann rearrangement was exploited to construct the 3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-one core. The flurobenzoxazepinone precursor 4a was then either inserted with pyridyl groups with or without a 4-methyl group resulting in compounds 5b and 5a, respectively, or transformed into N, N-dimethylamino intermediate 4b, which was consequently introduced with various substituted pyridyl moieties leading to final compounds 5c-5s under the catalysis of CuI. Similarly, the F group in compound 5b was replaced with amines in different sizes and capability in forming H-bond yielding compounds 5u-5z.

|
Download:
|
| Scheme 2. The synthesis of pyridyl benzoxazepinones. Reagents and conditions: (i) methyl 3-bromopropanoate, K2CO3, THF, 70 ℃, 16 h.; (ii) LiOH, H2O, r.t., 5 h; (iii) trifluoroacetic anhydride, trifluoroacetic acid, r.t., 12 h; (iv) NH2OH·HCl, NaHCO3, EtOH, 90 ℃, 16 h; (v) polyphosphoric acid, 110 ℃, 4 h; (vi) General method A: corresponding bromopyridine, CuI, N, N-dimethyl-1, 2-ethanediamine, K2CO3, dioxane, 120 ℃, 16 h; (vii) General method B: corresponding amine, K2CO3, DMSO, 100 ℃, 16 h. | |
The inhibition of these compounds against CYP11B2 was evaluated (Table 1) using V79 cells coexpressing hCYP11B2 and NADPH-P450 reductase. As 11β-hydroxylase (CYP11B1) is the crucial enzyme in glucocorticoid biosynthesis, whose inhibition leads to impairments in immune response and metabolism, the selectivity of synthesized compounds over CYP11B1 was scrutinized as a pivotal aspect of safety. Noteworthy is that it is a huge challenge in achieving selectivity between CYP11B1 and CYP11B2 due to a high homological identity of more than 93%, which means only 30 out of total 503 amino acids are different and they all locate outside of the binding site. The reference compound fadrazole was a very potent inhibitor of both enzyme with an IC50 of 15 nmol/L toward CYP11B2, however, exhibited a poor selectivity factor (SF=IC50 CYP11B1/IC50 CYP11B2) of 7.
|
|
Table 1 The inhibition of the designed benzoxazepinones against hCYP11B1 and hCYP11B2 in contrast to reference compound fadrazole.a |

{kind=link}
{kind=link}
{kind=link}
In accordance to our design strategy, a fluoro group was initially employed on the benzoxazepinone core to form an H-bond with Glu383, the resulting compounds 5a and 5b indeed strongly inhibited CYP11B2 with IC50s of 138 and 79 nmol/L, respectively, and good selectivity over CYP11B1 was achieved (SFs around 60). When F was replaced by an N, N-dimethylamino moiety, the inhibitory potency was augmented by about one-third leading to compounds 5c and 5l (IC50=102 and 52 nmol/L, respectively). Surprising is the elevation of selectivity factors by approximately two- to three-fold to 123 (5c) and 165 (5l). Due to these improvements observed, the N, N-dimethylamino group was sustained when the influence of the pyridyl part was investigated.
It has been clearly demonstrated that the 3-pyridyl analogues were more potent than the corresponding 4-pyridyl compounds by six- to twelve-fold, (for comparison, see compound pairs of 5c & 5d, 5e & 5f, 5g & 5h, and 5j & 5k), leaving 4-pyridyl compounds as weak CYP11B2 inhibitors with IC50s greater than 500 nmol/L. This observation indicated that coordination was not well-established between the 4-pyridyl sp2 hybrid N and the heme iron even if certain flexibility was granted by the twisted benzoxazepinone scaffold.
With regard to substituents on the pyridyl, obvious "ortho-effects" presented, where compounds with substituents adjacent to the benzoxazepinone core (i.e., at the 4′-position of the 3-pyridyl) were approximately a quarter more potent than the corresponding analogues with the same substituents at the 5′-postion (for comparison, see compound pairs of 5i & 5g, 5l & 5j, 5p & 5o, 5r & 5q, and 5t & 5s). Since these groups were varied in bulkiness and electrostatic properties, blocking free rotations of the pyridyl moiety and thus locking the molecule into a conformation preferred for CYP11B2 binding probably served as the predominate reason for the observed "ortho-effects".
Moreover, substituents on the pyridyl part significantly influenced the inhibitory potency with their capability in donating or attracting electrons as well. When being introduced at the 5′-position of 3-pyridyl, a methyl group increased the potency by around 25% to 75 nmol/L (5j) compared to the non-substituted one (5c, IC50=102 nmol/L). Another electron-donating group OMe (5m) rendered the compound even stronger (IC50=67 nmol/L), while Cl analogue 5g was similarly potent as the non-substituted 5c. In contrast, strong electron-withdrawn groups weakened CYP11B2 inhibition, as manifested by compounds 5n (acetyl, 139 nmol/L), 5o (COOMe, 162 nmol/L), 5q (CF3, 119 nmol/L), and 5s (CN, 134 nmol/L). The only exception is the fluoro compound 5e showing an IC50 of 65 nmol/L, which might indicate a potential H-bond between the F and the nearby Thr314. Such an observation was probably the consequence of distinct electron densities that could be exploited by the sp2 hybrid N to coordinate to the heme iron, which was evidently impacted by substituents with different electrostatic properties. A similar trend echoing this structure-activity relationship was also observed for compounds with substituents at the 4′-position of 3-pyridyl, where Me (5l, 52 nmol/L) > Cl (5i, 71 nmol/L) > CF3 (5r, 86 nmol/L) > CN (5t, 98 nmol/L) ≥ H (5c, 102 nmol/L) > COOMe (5p, 128 nmol/L). Importantly, the "ortho-effects" neutralized the negative influences of certain electron-withdrawn groups, leaving Cl and CF3 compounds (5i and 5r, respectively) more potent than the non-substituted 5c, while resulting CN analogue 5t in a comparable potency.
Due to the superior performance of 4′-methylpyridin-3-yl on both CYP11B2 inhibition and selectivity (5l: IC50=52 nmol/L, SF=165), this moiety was preserved when modifying substituents on the benzoxazepinone core. Guided by the docking study, optimizations were focused on the 8-position, where substituents were expected to form various interactions with Glu383 and Met111 (Fig. 1). Encouraged by the improvements of inhibitory potency observed when a fluoro group was replaced by an N, N-dimethylamino moiety, further amino substituents were inserted. Bulky pyrrolidinyl (5u) and piperidinyl (5v) reduced the potency by approximate three-fold (IC50s around 150 nmol/L) compared to the N, N-dimethylamino analogue 5l, probably due to potential steric clashes. Nevertheless, those negative impacts were compensated by introducing further H-bond forming groups in closer proximity to the Glu383 and/or Met111 (Fig. 1). H-bond accepting morpholino (5w) and N-methylpiperazinyl (5y) demonstrated stronger CYP11B2 inhibition with IC50 values of 39 and 25 nmol/L, respectively. N-acetylpiperazinyl (5z), however, resulted in reduced potency (IC50=87 nmol/L) even if another H-bond acceptor presented (carbonyl). Striking is that piperazinyl compound 5x, with a free secondary amino group at the far-end acting as an H-bond donor and acceptor, manifested a three-fold elevation of CYP11B2 inhibition (IC50=12 nmol/L) compared to the O analogue (5w, IC50=39 nmol/L) presenting only an H-bond acceptor at the same position. Besides H-bonds between the piperazinyl group and Glu383 as well as Met111, the salt bridge between the protonated amino moiety and Glu383 probably accounted for the increase of binding affinity.
In contrast to the poor selectivity of the reference compound fadrazole (SF=7), all derivatives exhibited good selectivity over CYP11B1 with SFs of more than 50. Amongst them, six compounds (5j, 5l, 5m 5w, 5x, and 5y) exhibited both potent CYP11B2 inhibition (IC50 < 100 nmol/L) and excellent selectivity over CYP11B1 (SF > 100). Compound 5x distinguished itself as the most promising one with an IC50 of 12 nmol/L and a superior selectivity factor of 157.
Furthermore, CYP17 and CYP19 are crucial enzymes in the biosynthesis of androgens and estrogens, respectively; while hepatic CYP enzymes involve in the metabolism of exogenous substances, in particular drugs. The inhibition of these enzymes leads to risks of drug-drug interactions and potential toxicities, a drug candidate of CYP11B2 inhibitor should therefore avoid interference with them. Compound 5x showed no significant inhibition against steroidogenic CYP17, CYP19 and a panel of hepatic CYP enzymes, including CYP1A2, CYP2C9, CYP2C19, CYP3A4, CYP2D6, and CYP2E1 (IC50s > 10 μmol/L).
To investigate the pharmacokinetic properties of compound 5x, it was subsequently applied to adult Wistar rats intravenously or per oral with doses of 5 or 25 mg/kg body weight, respectively. This compounds manifested good exposure via both application routes with areas-under-the-curve of around 45, 000 μg L-1 h and half-lives of 4 h to 5 h, which lead to a satisfactory bioavailability of 18.5% (Table S1 in Supporting information).
In conclusion, based on our previously developed CYP11B2 inhibitors, a series of pyridyl substituted 3,4-dihydrobenzo[f][1,4]oxazepin-5(2H)-ones were designed via a structure-based approach taking advantages of additional H-bonds and salt bridges with Glu383 and/or Met111. Six potent (IC50 < 100 nmol/L) and selective (SF > 100) CYP11B2 inhibitor (5j, 5l, 5m 5w, 5x, and 5y) were thus identified. As the most promising compound, 5x exhibited an IC50 of 12 nmol/L and a superior selectivity factor of 157, which is not only more potent than the reference fadrazole (IC50=21 nmol/L), but also 22-fold more selective. Importantly, 5x showed no significant inhibition against steroidogenic CYP17, CYP19 and a panel of hepatic CYP enzymes indicating an outstanding safety profile. As it manifested satisfactory pharmacokinetic properties in animals, compound 5x was considered as a drug candidate for further development.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis publication was supported by the National Natural Science Foundation of China (No. 81872739), the Zhujiang Distinguish Professorship of Guangdong Province, China (2018), the International Scientific Collaboration Program of Guangdong Province, China (No. 2020A0505100053) and the Key Research and Development Program of Guangdong Province, China (No. 2019B02021002).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.021.
| [1] |
J.M. Luther, Curr. Opin. Nephrol. Hypertens. 25 (2016) 16-21. DOI:10.1097/MNH.0000000000000189 |
| [2] |
A. Cannavo, A. Elia, D. Liccardo, G. Rengo, W.J. Koch, Vitam. Horm. 109 (2019) 387-406. DOI:10.1016/bs.vh.2018.09.005 |
| [3] |
A. Shrestha, R.C. Che, A.H. Zhang, Adv. Exp. Med. Biol. 1165 (2019) 325-346. DOI:10.1007/978-981-13-8871-2_15 |
| [4] |
F. Zannad, W. Gattis Stough, P. Rossignol, et al., Eur. Heart J. 33 (2012) 2782-2795. DOI:10.1093/eurheartj/ehs257 |
| [5] |
N. Wan, A. Rahman, A. Nishiyama, J. Hum. Hypertens. 35 (2021) 148-156. DOI:10.1038/s41371-020-0377-6 |
| [6] |
R. Gros, Q. Ding, L.A. Sklar, et al., Hypertension 57 (2011) 442-451. DOI:10.1161/HYPERTENSIONAHA.110.161653 |
| [7] |
M. Packer, Circulation 137 (2018) 1614-1631. DOI:10.1161/circulationaha.117.032474 |
| [8] |
Q. Hu, L. Yin, R.W. Hartmann, J. Med. Chem. 57 (2014) 5011-5022. DOI:10.1021/jm401430e |
| [9] |
M. Voets, I. Antes, C. Scherer, et al., J. Med. Chem. 48 (2005) 6632-6642. DOI:10.1021/jm0503704 |
| [10] |
L. Yin, Q. Hu, J. Emmerich, et al., J. Med. Chem. 57 (2014) 5179-5189. DOI:10.1021/jm500140c |
| [11] |
Q. Hu, L. Yin, A. Ali, et al., J. Med. Chem. 58 (2015) 2530-2537. DOI:10.1021/acs.jmedchem.5b00079 |
| [12] |
N. Strushkevich, A.A. Gilep, L. Shen, et al., Mol. Endocrinol. 27 (2013) 315-324. DOI:10.1210/me.2012-1287 |