 2021, Vol. 32
2021, Vol. 32 


Copper catalyzed carboboration of alkenes via borocupration across C-C π bond and subsequent electrophilic capture of the in situ formed organocuprate intermediate by a broad range of carbon based electrophiles has emerged as an important means for difunctionalization of alkenes [1-17], which leads to versatile and valuable organoboron compounds [18]. In particular, when a suitable C-electrophile is linked to the targeting alkene moiety properly, a cyclization will occur giving rise to a borylated (hetero)cyclic molecule [19-27], as exemplified by the elegant constructions of silylcyclopropylboronates from γ-silylallylic carbonates [28] and cyclobutylboronates from homoallylic sulfonates reported by Ito and Sawamura group [29, 30]. Following this strategy, we [31, 32] and others [33-35] have recently developed diastereo- and/or enantioselective synthesis of borylated 2, 3-disubstituted indolines 2 and 2, 3, 4-trisubstituted tetrahydroquinolines 3 and 3′ from alkenyl aldmines 1a and 1b respectively (Scheme 1a). In 2018, Lautens and colleagues reported a borylative cyclization of 2-vinylaniline carbamoyl chlorides 4, delivering biologically important 3, 3-disubstituted oxindoles 5 (Scheme 1b) [36]. In line with our interest in this field, we proposed that regioselective borylative cyclization of arylallyl carbamoyl chlorides 6 would provide a straightforward access to highly desired 2-aryl-3-boryl-γ-lactams 7 (Scheme 1c) [37], as the γ-lactam is a common core structure found in various natural products and synthetic molecules of biological interests [38-45].

|
Download:
|
| Scheme 1. (a) Borylative cyclization of 2-vinylanilinyl aldmines; (b) Borylative cyclization of 2-vinylaniline carbamoyl chlorides; (c) Borylative cyclization of allyl carbamoyl chlorides. | |
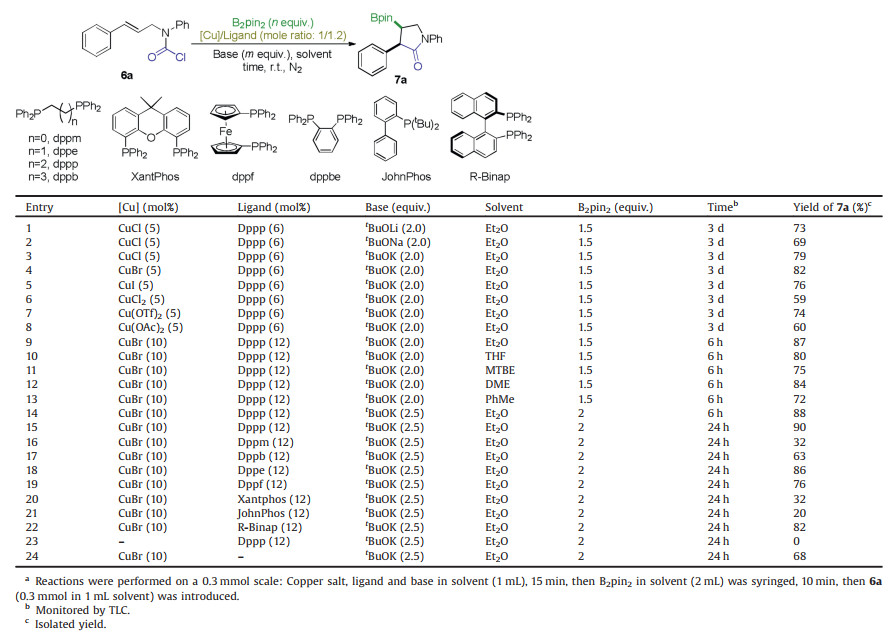
Our studies were commenced with cinnamyl carbamoyl chloride 6a, made from corresponding cinnamyl amine. Extensive catalyst-ligand screening and condition optimization were carried out, and selected data were tabulated in Table 1. It was found that premixing CuCl (5 mol%), Dppp (6 mol%) and tBuOLi (2.0 equiv.) in diethyl ether forms a homogenous solution, subsequent introduction of B2pin2 (1.5 equiv.) and substrate 6a (1.0 equiv.) affords a slow reaction which fails to achieve full conversion even after 3 days. Pleasingly, 3-boryl-2-phenyl γ-lactam 7a was collected in 73% yield as a single diastereomor (entry 1). The relative stereochemistry of the 2, 3-disubstituted lactam was determined as cis-configuration by single crystal X-Ray crystallography (Fig. 1). tBuOK as the base gave a noticeable higher yield (79%) than tBuOLi (73%) and tBuONa (69%) did in the same conditions (entry 3 vs. entries 1 and 2). Examination of a diverse copper salts revealed that CuBr performed best as a pre-catalyst (entries 4–8). Doubling the catalyst/ligand loading resulted in a further enhancement in yield to 87% even in a much shorter reaction time of only 6 h (entry 9). Brief solvent screening showed that THF and dimethoxylethane (DME) are similarly good media for this reaction, giving better results than methyl tert-butyl ether (MTBE) and toluene did (entries 10–13). However, at this stage, conditions capable to realize complete consumption of 6a for sure were not identified yet. Increasing the B2pin2/tBuOK combination from equivalents of 1.5/2.0 to 2.0/2.5 in conjunction with elongating the reaction time to 24 h effected full conversion and pushed the yield up to 90% (entries 14 and 15). With the so far optimal conditions, we progressed to evaluate ligand impact on the reaction (entries 16–22). It was found that dppe and R-Binap are as excellent as dppp; dppb and dppf are also viable but with moderate yields; and dppm, Xantphos and JohnPhos are much inferior ligands for this copper catalyzed process. These data evidenced the important role of ligand (entries 16–22). Without copper salt, the reaction didnot take place at all, in support the metal catalytic pathway (entry 23). In the absence of a ligand, 7a was still obtained albeit in a lower yield (entry 24), a result not out of expectation in the mechanistic point of view.
|
|
Table 1 Reaction conditions optimization.a |
{kind=link}

|
Download:
|
| Fig. 1. The ORTEP drawing of 7a (CCDC: 2038413). | |
{kind=link}
It is argued that the borocupration is a syn addition process that affords the syn intermediate INT-6/7 solely. The subsequent interception of the C-Cu nucleophile by the pendent carbamoyl chloride proceeds in a configuration retention fashion to accomplish the cis product 7a exclusively (Scheme 2).

|
Download:
|
| Scheme 2. Explanation of the relative stereochemistry of borylative cyclization. | |
{kind=link}
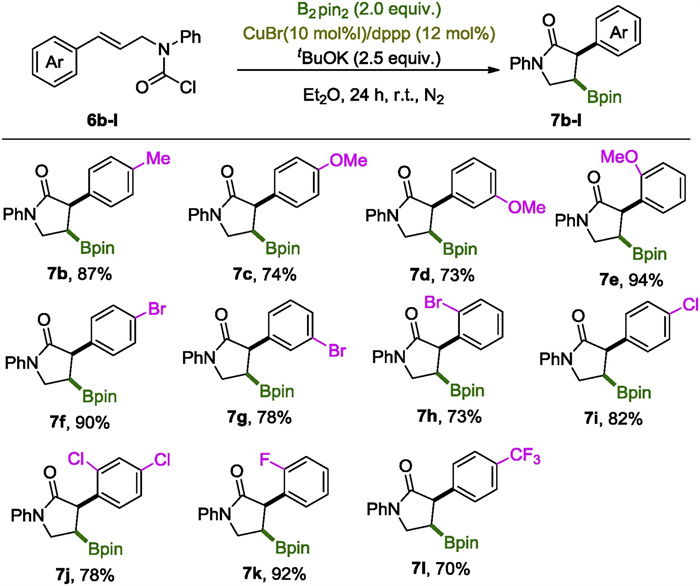
With the realization of the borylative cyclization reaction of 6a and the establishment of the optimal conditions (conditions used in entry 15 in Table 1), we set out to explore the substrate scope. Firstly, variation on the aromatic ring of the allyl segment was investigated, and the results were collected in Scheme 3. As expected 7b was obtained in high yield as a p-Me group on the phenyl group can scarcely change the steric and electronic properties of a substrate. Carbamoyl chlorides 6c-e, incorporating an electron releasing methoxyl group on the aromatic ring, were all nice substrates for this reaction to give 7c-e in > 73% yields, especially in the case of 7e, a counterintuitively excellent yield of 94% was obtained. Notably, transition metal vulnerable phenyl bromides were compatible with this copper catalyzed process as demonstrated by clean reactions of 6f-h, affording boryl lactams 7f-h in high to excellent yields. Mono and di-chlorinated substrates 6i and 6j underwent this carboboration process smoothly as well, yielding 7i and 7j in 82% and 78% yields respectively. Fluorinated boryl lactam 7k was obtained in excellent yield, indicating the HF elimination process, a common pathway promoted by strong alkoxide bases, did not interfere with the current reaction, probably due to the Lewis acidic boryl agents in the system that could mitigate the strong basicity. 6l with a 4-CF3 group was converted into lactam 7l smoothly.

|
Download:
|
| Scheme 3. The scope of the cinnamyl segments. Reactions were performed on a scale of 0.3 mmol; isolated yields were reported. | |
{kind=link}
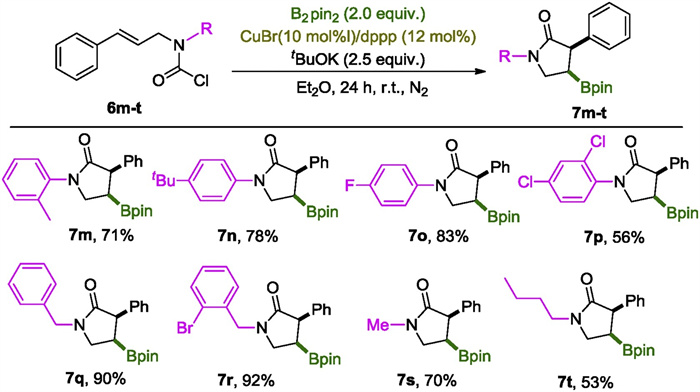
Next, the impact of N-substitution on the reaction was inspected (Scheme 4). As expected, 6m-o with aromatic substituents all proceed smoothly to give corresponding lactams 7m-o in high yields. 2, 4-Dichloanilline derived carbamoyl chloride 6p was transformed into 7p in modest 56% yield. Switching the N-aryl group to N-benzyl group improved the reaction prominently as evidenced by the excellent yields collected for 7q and 7r. N-Alkyl substitution was also applicable while moderate yields were obtained as demonstrated by the reactions of 6s and 6t.

|
Download:
|
| Scheme 4. The scope of the N-substituent. Reactions were performed on a scale of 0.3 mmol; isolated yields were reported. | |
{kind=link}
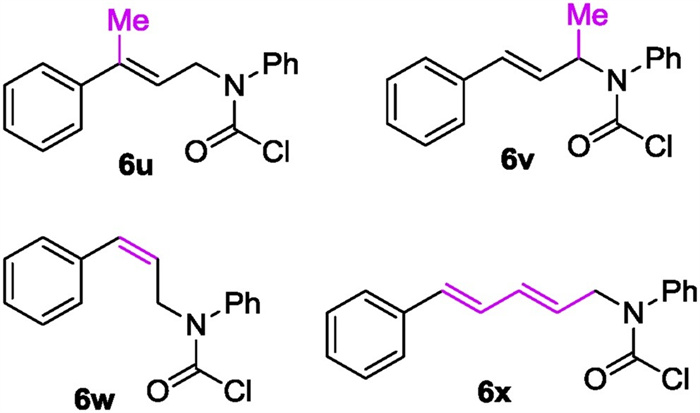
Expansion of this protocol to 6u, a tri-substituted allyl carbamoyl chloride that would give a 2, 2-disubstituted lactam if succeeds, resulted in an inert reaction as no conversion of the starting material was observed, presumably attributing to the difficult formation of a tertiary organocuprate and/or the following intramolecular coupling event. Relocating the methyl group to the allylic position furnishes 6v which was submitted to the reaction under optimal conditions as well. However the expected transformation did not happen even in refluxing ether. More surprisingly, Z-alkenyl substrate 6w did not undergo this process either. Moreover, dienyl substrate 6x did react under the same conditions, but a complex mixture was observed. These unsuccessful cases call for future studies. (Fig. 2)

|
Download:
|
| Fig. 2. Several yet unsuccessful substrates. | |
{kind=link}
Treatment with H2O2/NaH2PO4, 7k was converted to alcohol 8 with high efficiency (Scheme 5 top). Following the method developed in Argarwal laboratory [46], furan substituted lactam 9 was obtained successfully from 7f (Scheme 5 bottom). These reactions demonstrate the usefulness of this reaction and highlight the versatility of the boryl group for further transformation.

|
Download:
|
| Scheme 5. Examples of transformation of the borated products. | |
{kind=link}
In summary, an efficient construction of cis-2-aryl-3-boryl-γ-lactams has been achieved through copper catalyzed borylative cyclization of arylallyl carbamoyl chlorides. This reaction is highly diastereoselective giving cis-lactams exclusively. The limitations in substrate scope demand further studies and we would like to report the progress in due time.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors thank the National Natural Science Foundation of China (No. 21672027) for financial support. This work is also supported by High-Level Entrepreneurial Talent Team of Jiangsu Province (No. 2017-37).
| [1] |
F.J.T. Talbot, Q. Dherbassy, S. Manna, et al., Angew. Chem. Int. Ed. 59 (2020) 20278-20289. DOI:10.1002/anie.202007251 |
| [2] |
A. Whyte, A. Torelli, B. Mirabi, et al., ACS Catal. 10 (2020) 11578-11622. DOI:10.1021/acscatal.0c02758 |
| [3] |
W. Xue, M. Oestreich, ACS Cent. Sci. 6 (2020) 1070-1081. DOI:10.1021/acscentsci.0c00738 |
| [4] |
Z. Liu, Y. Gao, T. Zeng, et al., Isr. J. Chem. 60 (2020) 219-229. DOI:10.1002/ijch.201900087 |
| [5] |
D. Hemming, R. Fritzemeier, S.A. Westcott, et al., Chem. Soc. Rev. 47 (2018) 7477-7494. DOI:10.1039/c7cs00816c |
| [6] |
Y. Shimizu, M. Kanai, Tetrahedron Lett. 55 (2014) 3727-3737. DOI:10.1016/j.tetlet.2014.05.077 |
| [7] |
K.F. Zhuo, W.Y. Xu, T.J. Gong, et al., Chem. Commun. 56 (2020) 2340-2343. DOI:10.1039/c9cc08485a |
| [8] |
Z. Su, Y. Feng, R. Zou, et al., Chem. Commun. 56 (2020) 7483-7486. DOI:10.1039/c9cc09902f |
| [9] |
D. Fiorito, Y. Liu, C. Besnard, et al., J. Am. Chem. Soc. 142 (2020) 623-632. DOI:10.1021/jacs.9b12297 |
| [10] |
N.Y. Wu, X.H. Xu, F.L. Qing, ACS Catal. 9 (2019) 5726-5731. DOI:10.1021/acscatal.9b01530 |
| [11] |
S.J. He, B. Wang, X. Lu, et al., Org. Lett. 20 (2018) 5208-5212. DOI:10.1021/acs.orglett.8b02155 |
| [12] |
B. Chen, P. Cao, Y. Liao, et al., Org. Lett. 20 (2018) 1346-1349. DOI:10.1021/acs.orglett.7b03860 |
| [13] |
L. Wen, H. Zhang, J. Wang, et al., Chem. Commun. 54 (2018) 12832-12835. DOI:10.1039/c8cc07032f |
| [14] |
F. Cheng, W. Lu, W. Huang, et al., Chem. Sci. 9 (2018) 4992-4998. DOI:10.1039/C8SC00827B |
| [15] |
T. Itoh, Y. Kanzaki, Y. Shimizu, et al., Angew. Chem. Int. Ed. 57 (2018) 8265-8269. DOI:10.1002/anie.201804117 |
| [16] |
H. Yoshida, I. Kageyuki, K. Takaki, Org. Lett. 15 (2013) 952-955. DOI:10.1021/ol4001526 |
| [17] |
H. Ito, Y. Kosaka, K. Nonoyama, et al., Angew. Chem. Int. Ed. 47 (2008) 7424-7427. DOI:10.1002/anie.200802342 |
| [18] |
J. Lv, B. Zhao, Y. Han, et al., Chin. Chem. Lett. 32 (2021) 691-694. DOI:10.1016/j.cclet.2020.06.028 |
| [19] |
C.Y. He, Q.H. Li, X. Wang, et al., Adv. Synth. Catal. 362 (2020) 765-770. DOI:10.1002/adsc.201901108 |
| [20] |
J. Sendra, R. Manzano, E. Reyes, et al., Angew. Chem. Int. Ed. 59 (2020) 2100-2104. DOI:10.1002/anie.201913438 |
| [21] |
A. Whyte, A. Torelli, B. Mirabi, et al., Org. Lett. 21 (2019) 8373-8377. DOI:10.1021/acs.orglett.9b03144 |
| [22] |
A. Whyte, B. Mirabi, A. Torelli, et al., ACS Catal. 9 (2019) 9253-9258. DOI:10.1021/acscatal.9b03216 |
| [23] |
P. Zheng, X. Han, J. Hu, et al., Org. Lett. 21 (2019) 6040-6044. DOI:10.1021/acs.orglett.9b02199 |
| [24] |
J. Royes, S. Ni, A. Farre, et al., ACS Catal. 8 (2018) 2833-2838. DOI:10.1021/acscatal.8b00257 |
| [25] |
D. Li, Y. Park, J. Yun, Org. Lett. 20 (2018) 7526-7529. DOI:10.1021/acs.orglett.8b03286 |
| [26] |
S. Zhou, F. Yuan, M. Guo, et al., Org. Lett. 20 (2018) 6710-6714. DOI:10.1021/acs.orglett.8b02801 |
| [27] |
A.R. Burns, J. Solana Gonzalez, H.W. Lam, Angew. Chem. Int. Ed. 51 (2012) 10827-10831. DOI:10.1002/anie.201205899 |
| [28] |
C. Zhong, S. Kunii, Y. Kosaka, et al., J. Am. Chem. Soc. 132 (2010) 11440-11442. DOI:10.1021/ja103783p |
| [29] |
K. Kubota, E. Yamamoto, H. Ito, J. Am. Chem. Soc. 135 (2013) 2635-2640. DOI:10.1021/ja3104582 |
| [30] |
H. Ito, T. Toyoda, M. Sawamura, J. Am. Chem. Soc. 132 (2010) 5990-5992. DOI:10.1021/ja101793a |
| [31] |
Y.P. Bi, H.M. Wang, H.Y. Qu, et al., Org. Biomol. Chem. 17 (2019) 1542-1546. DOI:10.1039/c8ob03195a |
| [32] |
H.M. Wang, H. Zhou, Q.S. Xu, et al., Org. Lett. 20 (2018) 1777-1780. DOI:10.1021/acs.orglett.8b00213 |
| [33] |
E.M. Larin, J. Loup, I. Polishchuk, et al., Chem. Sci. 11 (2020) 5716-5723. DOI:10.1039/d0sc02421j |
| [34] |
G. Zhang, A. Cang, Y. Wang, et al., Org. Lett. 20 (2018) 1798-1801. DOI:10.1021/acs.orglett.8b00246 |
| [35] |
D.X. Li, J. Kim, J.W. Yang, et al., Chem. Asian J. 13 (2018) 2365-2368. DOI:10.1002/asia.201800121 |
| [36] |
A. Whyte, K.I. Burton, J. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 13927-13930. DOI:10.1002/anie.201808460 |
| [37] |
A. Torelli, A. Whyte, I. Polishchuk, et al., Org. Lett. 22 (2020) 7915-7919. DOI:10.1021/acs.orglett.0c02837 |
| [38] |
J. Caruano, G.G. Muccioli, R. Robiette, Org. Biomol. Chem. 14 (2016) 10134-10156. DOI:10.1039/C6OB01349J |
| [39] |
L.W. Ye, C. Shu, F. Gagosz, Org. Biomol. Chem. 12 (2014) 1833-1845. DOI:10.1039/C3OB42181C |
| [40] |
Y. Park, S. Chang, Nat. Catal. 2 (2019) 219-227. DOI:10.1038/s41929-019-0230-x |
| [41] |
D. Zhou, C. Wang, M. Li, et al., Chin. Chem. Lett. 29 (2018) 191-193. DOI:10.1016/j.cclet.2017.06.007 |
| [42] |
S.Y. Hong, Y. Park, Y. Hwang, et al., Science 359 (2018) 1016-1021. DOI:10.1126/science.aap7503 |
| [43] |
Y.Y. Xu, A.R. Qian, X.F. Cao, et al., Chin. Chem. Lett. 27 (2016) 703-706. DOI:10.1016/j.cclet.2016.01.040 |
| [44] |
Z. Li, L. Song, C. Li, J. Am. Chem. Soc. 135 (2013) 4640-4643. DOI:10.1021/ja400124t |
| [45] |
M. Wasa, K.M. Engle, J.-Q. Yu, J. Am. Chem. Soc. 132 (2010) 3680-3681. DOI:10.1021/ja1010866 |
| [46] |
A. Bonet, M. Odachowski, D. Leonori, et al., Nat. Chem. 6 (2014) 584-589. DOI:10.1038/nchem.1971 |