 2021, Vol. 32
2021, Vol. 32 


b AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Kyoto 606-8501, Japan
Driven by the rapid consumption of fossil fuels, developing efficient generation technologies of clean and renewable energy becomes a top priority [1, 2]. In this regards, electrochemical water splitting to hydrogen gas, metal-air batteries, reversible fuel cells, and electrolyzers are hopeful green routes to energy storage and conversion [3, 4]. Nevertheless, a limiting factor hindering these technologies is the essential and sluggish reaction: the oxygen evolution reaction (OER, 4OH− → O2 + 2H2O + 4e−), which suffers from 4 steps including proton couple, electron transfer and breaking O-H bonds to form O=O bonds [5, 6]. Currently, benchmarking electrocatalysts for OER commonly are noble metal oxides such as RuO2 and IrO2 [7]. On this account, developing cost-effective OER electrocatalysts is of great significance [8, 9]. First-row transition-metal oxides are considered as promising alternatives to noble-metal based materials as OER catalysts due to their high abundance and regulable electronic structures for ion/intermediate absorption [10, 11].
Cu-based materials have attracted wide attention as electrocatalysts due to their rich reserves, non-toxicity and controllable micro/nanostructures [12-14]. However, the performances of most Cu-based electrocatalysts for OER are still far from satisfactory [15, 16]. One of the inherent reasons is that the high filling of anti-bonding state of Cu d band (3d104s1) makes it difficult to hybridize with the O 2p band of oxygen intermediates during the adsorption process [17, 18]. Hence, the key points to increase efficiency of Cu-based electrocatalysts are exploiting electronic structure changes, surface defects and hierarchical nanostructures to enhance the exposure of active sites [19-21]. Nonetheless, in order to realize the above-mentioned points, most of the reported synthetic strategies for highly efficient Cu-based electrocatalysts are complicated and of low yield [22, 23]. Intelligently, employing well-chosen metal-organic complexes/frameworks as self-sacrificing precursors can acquire metal oxides with expected proportions of each component and adjustable ordered structures [24, 25]. In our previous work [26], Co-alanine complexes were synthesized by chemical precipitation, of which the subsequent controllable calcination produces a novel porous nanostructure of N-doped CoOx showing a high and stable OER activity. More importantly, during the calcination of metal-organic complexes, the coordination bonds are broken to form edge-plane defects, C and N from the ligand are doped as heteroatoms, and the resulted oxide crystals grow in certain preferred orientation, all of which are of significance for enhancing the OER activities [27].
On the above basis, we report a facile synthetic method for CuO electrocatalysts with hierarchical nanostructures through chemical precipitation at room temperature and calcination in air. By investigating a series of products obtained at different calcination temperatures, the relationship between OER activities and the material characteristics such as specific surface areas, crystal growth orientations, and element components are demonstrated. The CuO-5 sample, obtained by calcination at 500 ℃, exhibits the smallest overpotentials of 290 mV in 1.0 mol/L KOH with a glassy carbon electrode (GCE) substrate for electrocatalyzing OER. Moreover, through the characterizations of CuO electrocatalysts after OER activities, the possible catalytic mechanism of CuO electrocatalysts and the influence factors of their OER performance are discussed.
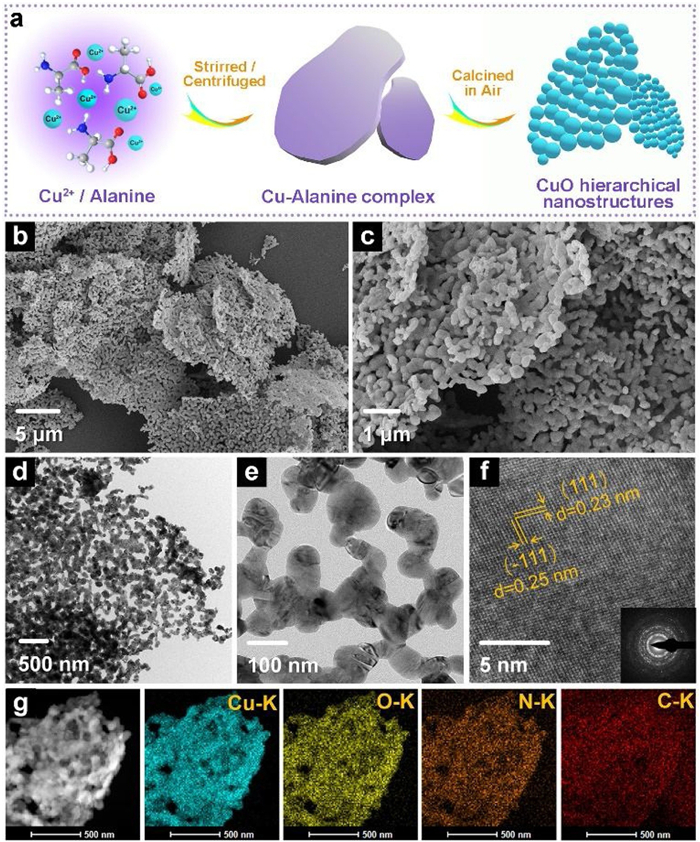
The synthetic route for CuO electrocatalyst is shown in Fig. 1a, and the description of detailed process is given in Supporting information. The calcination products obtained at 400, 500 and 600 ℃ are denoted as CuO-4, CuO-5 and CuO-6, respectively. Fig. S1 (Supporting information) exhibits the field emission scanning electron microscopy (FE-SEM) image of the precursor (Cu-alanine complex). It is observed that the complex has a micro-scale plate morphology. The powder X-ray diffraction (XRD) pattern of Cu-alanine complex (Fig. S2 in Supporting information) is in good agreenment with the previous report [28], illustrating the crystal structure of Cu-(ala)2·H2O. Fig. S3 (Supporting information) exhibits the Fourier transform infrared (FT-IR) spectrum of the precursor, in which the stretching modes of 1650–1100 cm−1 are attributed to the carboxylate, and the band at ~3222 cm−1 is characteristic of N-H vibration. Fig. S4 (Supporting information) shows the thermogravimetric (TG) curve of the precursor calcined in air atmosphere; the complex begins to lose the weight at ~210 ℃ and completes the decomposition at ~330 ℃ with 22.9% mass retained.

|
Download:
|
| Fig. 1. (a) Schematic illustration for the synthetic process of CuO electrocatalysts, and morphological characterizations of the CuO-5 sample: (b, c) FE-SEM images, (d, e) TEM images, (f) HRTEM image and SAED pattern (inset), and (g) STEM image and corresponding EDS mapping images (cyan-Cu-K, yellow-O-K, orange-N-K, red-C-K). | |
{kind=link}
FE-SEM and transmission electron microscopy (TEM) images of CuO-5 (Figs. 1b–e) reveal a hierarchical nanostructure of micro-scale plate assembled with nanoparticles, which can facilitate the ion contact with electrolyte and the expose of active sites. In addition, the high-resolution TEM (HRTEM) image in Fig. 1f indexes the lattice fringes of 0.23 and 0.25 nm to main crystal faces (111) and (111) of CuO, respectively. The diffraction rings in the selected area electron diffraction (SAED) pattern (inset of Fig. 1f) indicate the polycrystalline features. In Fig. 1g, energy-dispersive spectroscopy (EDS) elemental mapping images demonstrate the uniform distribution of Cu, O, N and C. The specific contents of C and N obtained by elemental analysis are summarized in Table S1 (Supporting information). The corresponding microscopic images of CuO-4 and CuO-6 are exhibited in Figs. S5 and S6 (Supporting information), respectively. With increasing the calcination temperature, the size of nanoparticles increases, while the contents of C and N decrease. The N2 adsorption-desorption isotherms of CuO electrocatalysts (Fig. S7 in Supporting information) reveal that the BET surface areas, calculated by the Barrett-Joyner-Halenda method, of CuO-4, CuO-5 and CuO-6 are 10.352, 10.591, and 9.065 m2/g, respectively.
The XRD patterns of CuO-4, CuO-5 and CuO-6 are shown in Fig. 2a. The characteristic peaks coincide well with the monoclinic CuO phase (JCPDS No. 80-1916). Significantly, contrast to the peak (111) at ~38.6°, the intensity of peak (111) at ~35.5° increases with increasing the calcination temperature, as the fusion of CuO nanoparticles at high temperatures minimizes the total energy by exposing the most stable facet with the lowest surface energy. The Raman spectra (Fig. S8 in Supporting information) of CuO electrocatalysts display a broad band at 298 cm−1 originating from the Ag phonon mode of Cu-O. The valence states of Cu, O, C, N elements of CuO electrocatalysts are analyzed by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2b, the high-resolution spectrum of Cu 2p valence state is characteristic of CuO materials where the main peaks at 932.8 eV and 952.6 eV are attributed to Cu 2p3/2 and Cu 2p1/2, along with the satellite peaks at 940.2/942.8 eV and 960.3/961.4 eV, respectively. Correspondingly, in the O 1s spectrum (Fig. 2c), the peak at 528.8 eV is ascribed to the Cu-O bond of lattice oxygen, and the peak at 530.4 eV is attributed to the oxygenic groups adsorbed on the surface of CuO particles. In C 1s spectrum of CuO (Fig. 2d), the peaks at 283.7, 284.8 and 287.4 eV are ascribed to the Cu-C, graphite C-C, and Cu-C-N, respectively. Generally, C or N doping on catalyst surface contributes to increase the efficiency of the OER. However, XPS measurements show no signal of N, and weak peaks of C, suggesting that little N and C exist on the surface of CuO nanoparticles. This may be due to the high temperature treatment in air atmosphere, which accelerates the oxidization and the loss of N and C existing on the surface of CuO nanoparticles. Therefore, in this work, it is difficult to quantitatively discuss the influence of contents of C and N for OER activities.

|
Download:
|
| Fig. 2. Characterizations of the CuO electrocatalysts: (a) XRD patterns, and XPS high-resolution spectra for (b) Cu 2p, (c) O 1s and (d) C 1s levels. | |
{kind=link}
The OER catalytic activities of CuO-4, CuO-5 and CuO-6 are evaluated by electrochemical measurements in a three-electrode system, and the details of electrode preparation and measurement parameters are given in electrochemical measurements. In the linear sweep voltammetry (LSV) curves of Fig. 3a, CuO-5 shows the earliest potential with a value of 1.522 V at a current density of 10 mA/cm2 (Ej=10 mA/cm2), namely a low overpotential of 290 mV. CuO-4 and CuO-6 show inferior catalytic activities, which require overpotentials of 330 and 370 mV, respectively, to reach the current density of 10 mA/cm2. Fig. 3b shows the Tafel slopes derived from the LSV curves, in which CuO-5 shows the expected lowest Tafel slope of 222.63 mV/dec, indicating the most favorable kinetics. In addition, the relative electrochemical surface areas (ECSAs) are estimated through the cyclic voltammograms (CV) curves with different scan rates at the non-faradaic potential window (Fig. S9 in Supporting information), of which more details of measurements and calculations are given in Supporting Information. As shown in Fig. 3c, the ECSAs of CuO-4, CuO-5 and CuO-6 are 6.03, 8.16 and 4.15 mF/cm2, respectively, demonstrating the highest surface roughness of CuO-5. Nyquist plots of Alternating-current (AC) impedance of the CuO electrocatalysts were measured versus Hg/HgO in 1.0 mol/L KOH (Fig. S10 in Supporting information). Compared with the other two electrocatalysts, the charge transfer interface of CuO-5 exhibits the least resistance and the fastest dynamics, in consistence with the results of Tafel slopes. Fig. S11 (Supporting information) shows the amperometric I-t curves in 1.0 mol/L KOH at a current density of 10 mA/cm2, where only a slight current decrease to 8.10 mA was observed after the test up to 4000 s, exhibiting a satisfactory cycle stability of the CuO-5 electrocatalysts.

|
Download:
|
| Fig. 3. Electrochemical test of CuO electrocatalysts: (a) LSV curves in 1.0 mol/L KOH electrolyte with a scan rate of 5 mV/s; (b) Tafel slope at J ≈ 10 mA/cm2 derived from the LSV curves; (c) ECSAs based on CV curves (Fig. S9) with scan rates of 10~100 mV/s. | |
{kind=link}
To further investigate the structural stability and the mechanism of catalytic action, a series of contrastive characterizations of CuO-5 before/after the CV tests were carried out. From the view of morphology, the hierarchical nanostructure of CuO-5 changes barely after 50 cycles of CV (Figs. S12a–c in Supporting information), while after 100 cycles (Figs. S12d and e in Supporting information) the CuO nanoparticles slightly agglomerate, and the lattice fringes in Fig. S12f (Supporting information) become irregular. Correspondingly, the XRD patterns of CuO-5 after 50/100 cycles of CV in Fig. 4a exhibit weaker peaks. In Fig. S12g (Supporting information), CuO-5 maintains the same element distribution before/after the CV tests. More significantly, the contrastive XPS measurements of the CuO-5 electrocatalysts (Figs. 4b–d) prove that CuO-5 largely retains its original components and the valence states after the electrochemical cycles.

|
Download:
|
| Fig. 4. Characterizations of CuO-5 as-prepared and after CV tests of 50 and 100 cycles: (a) XRD patterns and high-resolution XPS spectra of (b) Cu 2p, (c) O 1s and (d) C 1s levels. | |
{kind=link}
According to the previous reports, metal cations with high oxidation states usually serve as active sites for intermediate adsorption and electron transfer within the OER process [29]. Under the alkaline condition and impressed voltage, a part of active Cu2+ on the interface will be oxidized to Cu3+, while the corresponding anodic peak is unapparent due to the overlap with the OER current in LSV curves. In the CV curves of CuO-5 in Fig. S13 (Supporting information), the raised platform and the cathodic peak at ~1.35 V vs. RHE are attributed to the Cu2+ to Cu3+ oxidation and the Cu3+ to Cu2+ reduction, respectively. The electron transfers and intermediate adsorption during the OER process destroy the original regularity of monoclinic crystal of CuO. Nonetheless, the poor dynamics stability of the Cu3+ species makes it difficult to track the presence of Cu3+ through the characterizations after electrochemical reaction. This is the reason why the valence states of Cu species hardly change in XPS while the XRD characteristic peaks weakened after the electrochemical reactions. On the basis of the previous literature [30] and the above analysis, it is presented that the OER process consists of the steps of electron transfer and intermediate adsorption on the active site of Cu2+ or Cu3+:
M represents a Cu active site of Cu2+ or Cu3+
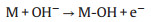
|
(1) |

|
(2) |
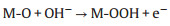
|
(3) |
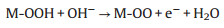
|
(4) |

|
(5) |
In summary, through a facile procedure of chemical precipitation at room temperature and calcination in air, CuO electrocatalysts with hierarchical nanostructures were obtained. Balancing the factors of specific surface area, crystal growth orientation, and element components affecting the catalytic activity, the product obtained at calcination temperature of 500 ℃ exhibits the smallest overpotentials of 290 mV in 1.0 mol/L KOH with GCE substrate. More importantly, based on a series of characterizations after electrocatalysis, the OER processes of electron transfer and intermediate adsorption with the active sites of Cu2+ and Cu3+ are discussed. Consequently, controllable calcination of Cu-alanine complexes to CuO electrocatalysts with hierarchical nanostructures provides a new venue to optimize the electronic structures, surface defects and hierarchical nanostructures for earth-abundant and efficacious electrocatalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (NSFC, Nos. U1904215, 21671170, 21673203, 21805192 and 21875207), the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), the Six Talent Plan (No. 2015-XCL-030), the Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515110735), China Postdoctoral Science Foundation (No. 2020M671612) and Jiangsu Province Postdoctoral Science Foundation (No. 2020Z082). Excellent Doctoral Dissertation of Yangzhou University and Undergraduate Scientific Research Innovation Projects in Jiangsu Province (No. 201911117036Z). We also acknowledge the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.037.
| [1] |
J. Mei, T. Liao, G.A. Ayoko, J. Bell, Z. Sun, Prog. Mater. Sci. 103 (2019) 596-677. |
| [2] |
J. Wu, S. Liu, F. Han, X. Yao, C. Wang, Adv. Mater. 7 (2020) 2000751. |
| [3] |
G. Chen, Y. Xu, L. Huang, A.I. Douka, B.Y. Xia, J. Energy Chem. 55 (2021) 183-189. DOI:10.1016/j.jechem.2020.07.012 |
| [4] |
S. Zhao, Y. Wang, J. Dong, et al., Nat. Energy 1 (2016) 16184. DOI:10.1038/nenergy.2016.184 |
| [5] |
D. Du, S. Zhao, Z. Zhu, F. Li, J. Chen, Angew. Chem. Int. Ed. 59 (2020) 18140-18144. DOI:10.1002/anie.202005929 |
| [6] |
S. Zheng, X. Guo, H. Xue, et al., Chem. Commun. 55 (2019) 10904-10907. DOI:10.1039/c9cc06113d |
| [7] |
X.W. (David) Lou, Angew. Chem. Int. Ed. 59 (2020) 18234-18239. DOI:10.1002/anie.202008129 |
| [8] |
S. Zheng, Y. Zheng, H. Xue, H. Pang, Chem. Eng. J. 395 (2020) 125166. DOI:10.1016/j.cej.2020.125166 |
| [9] |
Q. Li, N. Li, J. An, H. Pang, Inorg. Chem. Front. 7 (2020) 2089-2096. DOI:10.1039/d0qi00316f |
| [10] |
Y. Bai, G. Zhang, S. Zheng, et al., Sci. China Mater. 64 (2021) 137-148. DOI:10.1007/s40843-020-1342-2 |
| [11] |
X. Guo, Y. Xu, Y. Cheng, Y. Zhang, H. Pang, Appl. Mater. Today 18 (2020) 100517. DOI:10.1016/j.apmt.2019.100517 |
| [12] |
Z. Chen, T.J. Meyer, Angew. Chem. Int. Ed. 52 (2013) 700-703. DOI:10.1002/anie.201207215 |
| [13] |
J. Zhao, P.D. Tran, Y. Chen, et al., ACS Catal. 5 (2015) 4115-4120. DOI:10.1021/acscatal.5b00556 |
| [14] |
Z. Yin, D. Gao, S. Yao, et al., Nano Energy 27 (2016) 35-43. DOI:10.1016/j.nanoen.2016.06.035 |
| [15] |
G. Yuan, S. Yu, J. Jie, et al., Chin. Chem. Lett. 31 (2020) 1941-1945. DOI:10.1016/j.cclet.2019.12.034 |
| [16] |
N.K. Chaudhari, Y. Hong, B. Kim, S. Il Choi, K. Lee, J. Mater. Chem. A 7 (2019) 17183-17203. DOI:10.1039/c9ta05309c |
| [17] |
W. Huang, Q. Liu, Z. Zhou, et al., Nat. Commun. 11 (2020) 2312. DOI:10.1038/s41467-020-15965-8 |
| [18] |
F. Yu, F. Li, B. Zhang, H. Li, L. Sun, ACS Catal. 5 (2015) 627-630. DOI:10.1021/cs501510e |
| [19] |
Y. Zuo, Y. Liu, J. Li, et al., Chem. Mater. 31 (2019) 7732-7743. DOI:10.1021/acs.chemmater.9b02790 |
| [20] |
Z. Xiao, C. Xie, Y. Wang, R. Chen, S. Wang, J. Energy Chem. 53 (2020) 208-225. |
| [21] |
L. Yu, D. Deng, X. Bao, Angew. Chem. Int. Ed. 59 (2020) 15294-15297. DOI:10.1002/anie.202007604 |
| [22] |
X. Xiong, C. You, Z. Liu, A.M. Asiri, X. Sun, ACS Sustain. Chem. Eng. 6 (2018) 2883-2887. DOI:10.1021/acssuschemeng.7b03752 |
| [23] |
Y. Wang, S. Wang, D. Liu, et al., Chem. Commun. 56 (2020) 8750-8753. DOI:10.1039/d0cc03132a |
| [24] |
Z. Liang, R. Zhao, T. Qiu, R. Zou, Q. Xu, Energy Chem. 1 (2019) 100001. DOI:10.1016/j.enchem.2019.100001 |
| [25] |
D. Yan, Y. Li, J. Huo, et al., Adv. Mater. 29 (2017) 1606459. DOI:10.1002/adma.201606459 |
| [26] |
X. Li, J. Wei, Q. Li, et al., Adv. Funct. Mater. 28 (2018) 1800886. DOI:10.1002/adfm.201800886 |
| [27] |
X. Li, C. Wang, H. Xue, H. Pang, Q. Xu, Coord. Chem. Rev. 422 (2020) 213468. DOI:10.1016/j.ccr.2020.213468 |
| [28] |
R. Calvo, P.R. Levstein, E.E. Castellano, et al., Inorg. Chem. 30 (1991) 216-220. DOI:10.1021/ic00002a015 |
| [29] |
B. Zhang, L. Wang, Z. Cao, et al., Nat. Catal. 3 (2020) 985-992. DOI:10.1038/s41929-020-00525-6 |
| [30] |
Y. Deng, A.D. Handoko, Y. Du, S. Xi, B.S. Yeo, ACS Catal. 6 (2016) 2473-2481. DOI:10.1021/acscatal.6b00205 |