 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
MicroRNAs (miRNAs) are a class of short (19–25 nucleotides), protein non-coding RNAs that regulate gene expression by inducing messenger RNA (mRNA) degradation and translation inhibition. The abnormal expression of miRNA is associated with numerous biological processes and diseases [1, 2], particularly cancers [3, 4], heart disease [5-7], nervous system disease [8-10], and others [11-13]. In addition, the progression of disease is accompanied by the aberrant expression of multiple miRNAs. Therefore, the detection and monitoring of multiple miRNAs at the same time is useful for medical diagnosis. Compared to other RNA, miRNA has many unique characteristics, including a small size, low abundance, easy degradation, and sequence similarities among family members [14, 15]. However, the direct quantitative detection of multiple miRNAs is challenging. Hence, the development of sensitive and quantitative detection technologies has attracted significant attention.
MiRNA can be identified and quantified by many techniques. Most methods for multiple miRNAs detection are based on amplification or modification, such as quantitative real-time polymerase chain reaction (qRT-PCR) [16], microarray techniques [17], next generation sequencing (RNA-Seq) [18], and isothermal amplification [19]. Among these approaches, qRT-PCR and microarray are the conventional methods for the quantitative detection of miRNAs. qRT-PCR is the gold standard and is used to verify miRNA screening experiments, which require high precision and accuracy [20]. RNA-Seq is a high-throughput detection approach for profiling the transcriptome using deep-sequencing technologies [18] and has been the only platform to discover new miRNA. The isothermal-amplification-based methods have also been used to carry out multiple miRNAs analysis, and these are non-PCR amplification based methods [19, 21]. However, a key limitation is the complex probe design and the lack of suitable multiplex quantitative analysis methods. Further, amplification-based methods can suffer from quantitation bias and low fidelity. Amplification-free methods are generally less specific and sensitive than amplification-based methods. As a result, highly specific probes and highly sensitive instrumentation are required. Locked nucleic acids (LNA) and peptide nucleic acids (PNA) with high affinity and specificity and new nanomaterials are used for amplification-free detection of miRNA. For example, Metcalf et al. developed PNA-based fluorogenic biosensors based on an oligonucleotide template reaction for the detection of multiple miRNAs in body fluids [22]. In addition, a molybdenum disulfide (MoS2) integrated silica colloidal crystal barcode for multiplex miRNA screening that functions by measuring the fluorescence of quantum dots (QDs) has been prepared [23].
In the past decade, highly sensitive laser-induced fluorescence (LIF) detection systems have been used for the analysis of multiple miRNA sequences. The LIF system is one of the most sensitive spectral techniques, having a limit of detection as low as the yoctomole [24]. Using LIF in combination with capillary electrophoresis (CE), multiple miRNAs can be detected. For example, Jiang and co-workers developed an assay combining denaturing capillary gel electrophoresis with tandem adenosine-tailed DNA bridge-assisted splinted ligation to separate and detect miRNA in the Epstein–Barr virus [25]. In addition, Krylov et al. reported a method for the direct quantitative analysis of multiple miRNA sequences (DQAMmiR) using bare fused-silica capillary electrophoresis [26], and the design of LNA–DNA probes and the application of a dual-temperature technique allowed single base resolution to be achieved [27]. High-speed capillary sieving electrophoresis has also been reported to be an effective means for multiple miRNAs detection [28]. However, capillary liquid chromatography has excellent reproducibility and stability compared to those of CE. Further, with the miniaturization of liquid chromatography technology, ultranarrow bore channels have extensively used because of their very small sample volume requirements and high-separation efficiencies [29, 30]. In this study, a pico-HPLC-LIF detection system for the amplification-free determination of multiple miRNAs was established. A capillary having an inner diameter of 680 nm was used as the separation chromatographic column. Single stranded DNA (ssDNA) probes of different lengths complementary to the target miRNAs were designed for hybridization with the miRNAs, and the RNA-DNA hybrids were dyed with the YOYO-1 nucleic acid dye. By combining this with a highly sensitive LIF detection system, the separation and detection of multiple miRNAs with high sensitivity was achieved. The key to the chromatographic separation is hydrodynamic chromatography (HDC) and wall electrostatic interaction (WaLEI) separation.
A schematic of the pico-HPLC-LIF system for the detection and analysis of multiple miRNAs is presented in Fig. 1, along with an illustration of the separation mechanism. A diagram of the detection device is shown in Fig. S1 (Supporting information). The miRNAs were added to a 200 μL microcentrifuge tube with an excess of the ssDNA probe to ensure that most of the miRNAs was hybridized. Consequently, some redundant probes failed to bind to miRNAs (Fig. 1a). The YOYO-1 nucleic acid dye was added to the mixture after hybridization, which was then incubated at room temperature, resulting in fluorescent RNA-DNA hybrids (Fig. 1b) and non-fluorescent superfluous DNA probes. The dyed sample was injected into a capillary that had been washed with Tris–ethylenediaminetetraacetic acid (TE) buffer in advance, and then the hybrids was redistributed in the nanocapillary according to the molecular weight based on the HDC and WaLEI mechanism under nitrogen gas pressure-driven (Fig. 1c). This resulted in the redistribution of the hybrids to the detection window at the end of the nanocapillary for detection by the LIF system (Fig. 1d).

|
Download:
|
| Fig. 1. (a–d) Schematic illustration of detection and analysis for multiple miRNAs in the pico-HPLC-LIF system. (e) Schematic illustration of hydrodynamic chromatography model. (f) Mechanism of wall-layer electrostatic interactions. | |
{kind=link}
The separation mechanism of pico-HPLC is based on HDC and WaLEI [31, 32]. The HDC mechanism is shown in Fig. 1e. As shown, the velocity of a pressure-driven fluid in a nanocapillary has a parabolic profile because of hydrodynamic forces. Larger molecules have a greater hydrodynamic radius and move in the region of high velocity at the center of the channel; thus, the large molecules are eluted earlier than smaller molecules. Fig. 1f illustrates the WaLEI mechanism. When the pH of the solution is greater than 3, the silica hydroxy groups on the inner wall surface of the nanocapillary are dissociated, resulting in a negatively charged surface. RNA–DNA hybrids are negatively charged in solution, and electrostatic repulsion causes the RNA–DNA hybrids to move from the wall toward the center of the nanocapillary channel. The RNA-DNA hybrids with longer chain lengths have a greater negative charge, therefore, these longer chains experience greater electrostatic repulsion from the wall and are redistributed toward the high-velocity region at the center of the capillary. Thus, the longer hybrids elute earlier than the shorter ones.
The concentration and pH of the eluent buffer are significant parameters affecting the chromatographic behavior. Therefore, we studied the influence of the concentration and pH for RNA-DNA hybrids separation. Under pressure-driven conditions, the order of elution of the RNA-DNA hybrids is determined by the molecular weight. The ssDNA probe for mir-182 has a tag with the greatest drag, so the mir-182 hybrid is first to elute, followed by the hybrids of miR-155 and let-7a. The concentration of the
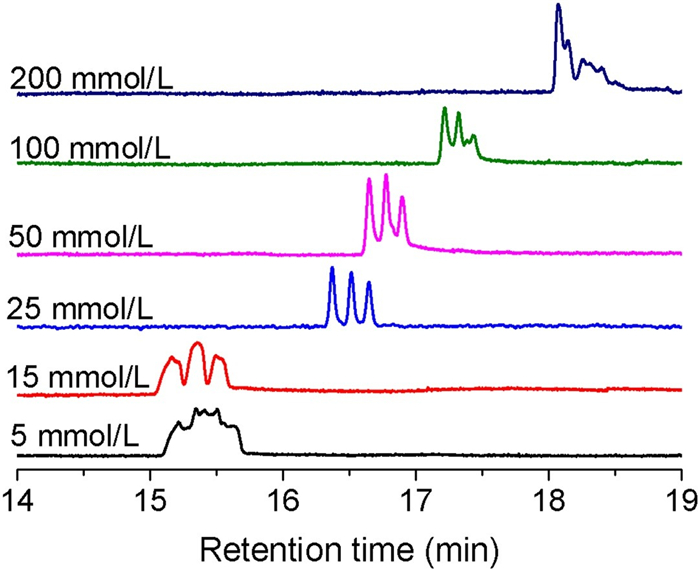
TE eluent also has a significant impact on the separation of RNA-DNA hybrids. Fig. 2 shows the separation chromatogram of the RNA-DNA hybrids at different TE concentrations (5–200 mmol/L). Under a constant driving pressure of 1300 psi, only TE concentrations of 15, 25 and 50 mmol/L yielded clearly resolved chromatographic peaks. Table S1 (Supporting information) lists the resolution and theoretical plates for all RNA-DNA hybrids at TE concentrations of 15, 25 and 50 mmol/L. Complete separation was achieved when the concentration of TE was 25 mmol/L, and the resolution was 2.0. The efficiencies of all hybrids were more than nine hundred thousand plates/meter, and more than one million plates/meter for let-7a hybrids has been achieved. When the TE concentration decreases, the electric double layer (EDL) on the capillary wall increases, so the radial migration of RNA-DNA hybrids is limited, and the effect of redistribution is weakened. However, the retention times of the hybrids increased and the separation effect decreased when the concentration of the TE eluent was greater than 25 mmol/L, which may be caused by the increase in solution viscosity and decrease in hydrodynamic effects at high concentrations.

|
Download:
|
| Fig. 2. Separation of a mixture of hybrids with different concentrations of TE buffer. The mixture of the hybrids contained three groups of DNA/miRNA. Peaks from left to right are mir-182, mir155 and let-7a. The concentration of the hybrids was 1.67 μmol/L, and the separation nanocapillary had an inner diameter of 680 nm and a total length of 50 cm (45 cm effective). The sample was injected at 200 psi for 20 s, and the separation was carried out at 1300 psi. | |
{kind=link}
As mentioned, the silica hydroxy groups on the inner wall of the capillary are ionized in aqueous solution, and the capillary wall is negatively charged. The eluent pH effects the dissociation efficiency of silica hydroxy groups on the capillary and affects the separation efficiency in capillary chromatography. Moreover, the stability of nucleic acid fragments is also affected by pH, especially that of the RNA-DNA hybrids. Therefore, it was necessary to identify the optimal pH of TE eluent to balance the ionization of silica hydroxy groups and then improve the reproducibility of sample migration. The degree of dissociation of the hydroxy groups increased rapidly with increase in pH above pH 3. Thus, to optimize the pH with respect to separation efficiency, seven different pH values (6.4, 7.0, 7.3, 7.6, 8.0, 8.3 and 8.6) of 25 mmol/L TE buffer were tested. In addition, each TE buffer was injected into the capillary 1 h before sample injection to balance the capillary. The separation results are shown in Fig. S3 (Supporting information), and the effect of the eluent pH on the resolution is depicted in Fig. S4 (Supporting information). The RNA-DNA hybrids could not be separated effectively when the eluent pH was less than 8.0, probably because of the lower dissociation of hydroxy groups under these conditions. The density of negative charge on the wall of the nanocapillary in acid solution is low, and the electrostatic interaction is relatively small. The electrostatic interaction is increased with the increasing of elute pH, so the resolution is improved. Highly alkaline conditions lead DNA to denature into ssDNA [33], so the experiment was not attempted at higher pH conditions. However, the separation efficiency of RNA-DNA hybrids reached baseline separation with little difference when the eluent pH was greater than or equal to 8.0. Considering that nucleic acids are easily denatured under highly alkaline condition, pH 8.0 TE buffer was selected for this work.
The effect of elution pressure (600–1400 psi) was investigated for RNA-DNA hybrids in a 48-cm long, 680-nm inner diameter capillary, and the separation results are shown in Fig. S5 (Supporting information). The retention time of the sample decreased proportionally with increase in elution pressure. The driving pressure and the volume velocity yielded a good linear correlation coefficient of 0.9937 (Fig. S6 in Supporting information). The resolution of adjacent RNA-DNA hybrids under different driving pressures was calculated, and the results are shown in Fig. S7 (Supporting information). The resolutions of RNA-DNA hybrids were markedly improved with increase in driving pressure, reaching a maximum at 1200 psi, after which they decreased slightly. The optimal elution pressured changed with changes in the experimental conditions, such as the composition of eluent, eluent pH, and channel size. To reduce the retention time, a higher elution pressure than that required for maximum elution efficiency is generally required (Fig. 3).

|
Download:
|
| Fig. 3. Chromatogram of a 26.04 nmol/L mixture of the RNA-DNA hybrids. The capillary had an internal diameter of 680 nm and a total length of 68 cm (effective length of 62 cm). The sample was injected at 200 psi for 20 s, and the separation was carried out at a pressure of 1500 psi. | |
{kind=link}
The limit of detection (LOD) was determined from the analysis of samples with known concentrations of the analyte and is the lowest detectable concentration. Typically, the LOD is calculated as three times signal-to-noise ratio (S/N ≥ 3). Thus, a sample was progressively diluted and analyzed. The injection time was 20 s at 200 psi, and the sample was driven at 1500 psi. Using an injection volume of 300fL, the LOD was 26.04 nmol/L (S/N ≈ 3.72) or 7.8 zmol (26.04 nmol/L × 300 fL). The volume of a single cell is typically picoliter in scale [34], so the sample volume requirement is less than that of a single cell. MiRNAs which is upregulated in cells was detected to more than 104 copies and the downregulated was less than 1000 copies [35]. Therefore, this approach can potentially be used to detect miRNAs in single cells.
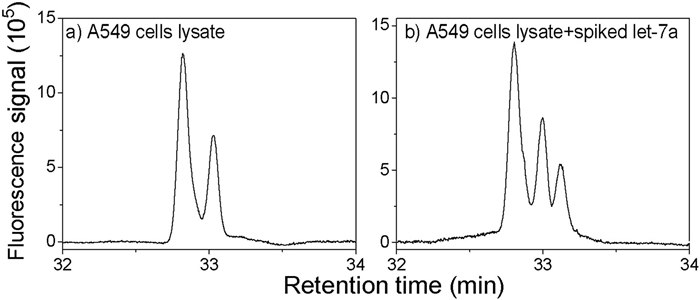
To test our method, we used a complex biological sample of miRNAs extracted from adenocarcinomic human alveolar basal epithelial (A549) cells. The ssDNA probes (5 μmol/L) were incubated with the complex miRNA sample for hybridization. Fig. 4 shows the separation results for the pure A549 cell lysate and the lysate spiked with let-7a. The concentration of spiked let-7a was 2.5 μmol/L and the final concentration of spiked let-7a was 230 nmol/L. Only two peaks were detected in the pure A549 cell lysate, whereas three peaks were detected from the sample of the lysate spiked with the let-7a. It can be inferred that two peaks of pure A549 cell lysate were mir-182 and mir-155, respectively, indicating that mir-182 and mir-155 were highly expressed in A549 cells. It is known that mir-182 and miR-155 are upregulated [36, 37] and let-7a is downregulated [38] in A549 cells; thus, our results are consistent with those of previous studies. These results illustrated that our method can be used for the detection and quantitative analysis of miRNA in complex samples.

|
Download:
|
| Fig. 4. Detection of multiple miRNAs from A549 cell extract: (a) 5 μmol/L of three ssDNA probes added to a sample of A549 cell extract and (b) 5 μmol/L of three ssDNA probes spiked with 2.5 μmol/L of let-7a and incubated with A549 cell extract. The nanocapillary had an internal diameter of 680 nm and was 65 cm long (60 cm effective length). The sample was injected at 200 psi for 20 s, and the separation was carried out at 1400 psi. | |
{kind=link}
In summary, we have demonstrated a pico-HPLC-LIF system for the amplification-free detection of multiple miRNAs. In addition, we have demonstrated the effects of eluent concentration, eluent pH, and driving pressure on the chromatographic behavior of the RNA-DNA hybrids. The optimal eluent concentration and pH were 25 mmol/L and 8.0, and the driving pressure was higher than 1200 psi. A nanocapillary having an inner diameter of 680 nm was used as a chromatographic column, and the RNA-DNA hybrids were separated via the HDC and WaLEI mechanisms. We found that a 300-fL sample was sufficient for separation and detection, and the limit of detection was 7.8 zmol or 4.7×103 molecules. The pico-HPLC-LIF strategy showed good performance in the simultaneous amplification-free detection of multiple miRNA sequences from complex A549 cells lysates, thus, a promising method for disease monitoring based on the detection of multiple miRNAs.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21625501, 21527808), the Beijing Outstanding Young Scientist Program (No. BJJWZYJH01201910005017).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.007
| [1] |
L. He, G.J. Hannon, Nat. Rev. Genet. 5 (2004) 522-531. DOI:10.1038/nrg1379 |
| [2] |
H. Dong, J. Lei, L. Ding, et al., Chem. Rev. 113 (2013) 6207-6233. |
| [3] |
N. Kosaka, H. Iguchi, T. Ochiya, Cancer Sci. 101 (2010) 2087-2092. DOI:10.1111/j.1349-7006.2010.01650.x |
| [4] |
B.P. Wijnhoven, M.Z. Michael, D.I. Watson, Br. J. Surg. 94 (2007) 23-30. DOI:10.1002/bjs.5673 |
| [5] |
J. Ooi, B. Bernardo, S. Singla, et al., RNA Biol. 14 (2017) 500-513. DOI:10.1080/15476286.2016.1181251 |
| [6] |
J.N.J. Buie, A.J. Goodwin, J.A. Cook, P.V. Halushka, H.K. Fan, Atherosclerosis 254 (2016) 271-281. DOI:10.1016/j.atherosclerosis.2016.09.067 |
| [7] |
S. Ikeda, S.W. Kong, J. Lu, et al., Physiol. Genomics 31 (2007) 367-373. DOI:10.1152/physiolgenomics.00144.2007 |
| [8] |
D.S. Greenberg, H. Soreq, Curr. Pharm. Design 20 (2014) 6022-6027. DOI:10.2174/1381612820666140314151924 |
| [9] |
O.C. Maes, H.M. Chertkow, E. Wang, H.M. Schipper, Curr. Genomics 10 (2009) 154-168. DOI:10.2174/138920209788185252 |
| [10] |
R. Fiore, G. Siegel, G. Schratt, BBA-Gene Regul. Mech. 1779 (2008) 471-478. |
| [11] |
D. Sekar, B.R. Shilpa, A.J. Das, Curr. Hypertens. Rep. 19 (2017) 57. DOI:10.1007/s11906-017-0752-z |
| [12] |
Y. Wei, A. Schober, Cell. Mol. Life Sci. 73 (2016) 3473-3495. |
| [13] |
M.E. Dumas, C. Emanueli, Diabetes 66 (2017) 565-567. DOI:10.2337/dbi16-0072 |
| [14] |
R.M. Graybill, R.C. Bailey, Anal. Chem. 88 (2016) 431-450. DOI:10.1021/acs.analchem.5b04679 |
| [15] |
K.A. Cissell, S. Shrestha, S.K. Deo, Anal. Chem. 79 (2007) 4754-4761. DOI:10.1021/ac0719305 |
| [16] |
S. Sharbati-Tehrani, B. Kutz-Lohroff, R. Bergbauer, J. Scholven, R. Einspanier, BMC Mol. Biol. 9 (2008) 34. DOI:10.1186/1471-2199-9-34 |
| [17] |
W. Li, K. Ruan, Anal. Bioanal. Chem. 394 (2009) 1117-1124. DOI:10.1007/s00216-008-2570-2 |
| [18] |
Z. Wang, M. Gerstein, M. Snyde, Nat. Rev. Genet. 10 (2009) 57-63. DOI:10.1038/nrg2484 |
| [19] |
J. Na, G.W. Shin, H.G. Son, S.J.V. Lee, G.Y. Jung, Sci. Rep. 7 (2017) 1-8. DOI:10.1038/s41598-016-0028-x |
| [20] |
P. Androvic, L. Valihrach, J. Elling, R. Sjoback, M. Kubista, Nucleic Acids Res. 45 (2017) e144. DOI:10.1093/nar/gkx588 |
| [21] |
S.C. Chapin, P.S. Doyle, Anal. Chem. 83 (2011) 7179-7185. DOI:10.1021/ac201618k |
| [22] |
G.A. Metcalf, A. Shibakawa, H. Patel, et al., Anal. Chem. 88 (2016) 8091-8098. DOI:10.1021/acs.analchem.6b01594 |
| [23] |
F.J. Bian, L.Y. Sun, L.J. Cai, et al., Biosens. Bioelectron. 133 (2019) 199-204. DOI:10.1016/j.bios.2019.02.066 |
| [24] |
W. Zhang, L. Liu, Q. Zhang, et al., Chem. Commun. 56 (2020) 2423-2426. DOI:10.1039/C9CC09594B |
| [25] |
R. Jiang, Y. Chang, S. Chen, et al., J. Chromatogr. A 1218 (2011) 2604-2610. DOI:10.1016/j.chroma.2011.02.061 |
| [26] |
D. Wegman, S. Krylov, Angew. Chem. Int. Ed. 50 (2011) 10335-10339. DOI:10.1002/anie.201104693 |
| [27] |
D. Wegman, F. Ghasemi, A. Stasheuski, et al., Anal. Chem. 88 (2016) 2472-2477. DOI:10.1021/acs.analchem.5b04682 |
| [28] |
W. Wang, X.Y. Cai, P. Lin, R.G. Bai, J. Sep. Sci. 41 (2018) 3925-3931. DOI:10.1002/jssc.201800635 |
| [29] |
R. Ishibashi, K. Mawatari, T. Kitamori, Small 8 (2012) 1237-1242. DOI:10.1002/smll.201102420 |
| [30] |
R. Li, Y. Shao, Y. Yu, X. Wang, G. Guo, Chem. Commun. 53 (2017) 4104-4107. DOI:10.1039/C7CC00799J |
| [31] |
L. Liu, V. Veerappan, Q. Pu, et al., Anal. Chem. 86 (2014) 729-736. DOI:10.1021/ac403190a |
| [32] |
X. Wang, L. Liu, G. Guo, et al., TrAC Trends Anal. Chem. 35 (2012) 122-134. DOI:10.1016/j.trac.2012.01.005 |
| [33] |
S. Howorka, Z. Siwy, Chem. Soc. Rev. 38 (2009) 2360-2384. DOI:10.1039/b813796j |
| [34] |
L. Lin, K. Mawatari, K. Morikawa, et al., Analyst 142 (2017) 1689-1696. DOI:10.1039/C7AN00220C |
| [35] |
B.J. Dodgson, A. Mazouchi, D.W. Wegman, C.C. Gradinaru, S.N. Krylov, Anal. Chem. 84 (2012) 5470-5474. DOI:10.1021/ac301546p |
| [36] |
F.L. Ning, F. Wang, M.L. Li, et al., Diagn. Pathol. 9 (2014) 143. DOI:10.1186/1746-1596-9-143 |
| [37] |
F. Liu, D.L. Song, Y.H. Wu, et al., Thorac. Cancer 8 (2017) 613-619. DOI:10.1111/1759-7714.12492 |
| [38] |
H.C. Jeong, E.K. Kim, J.H. Lee, et al., Mol. Med. Rep. 4 (2011) 383-387. |