 2021, Vol. 32
2021, Vol. 32 


b Department of Orthopaedics Trauma, Changhai Hospital, Second Military Medical University, Shanghai 200433, China;
c Institute of Biomedical and Pharmaceutical Technology, Fuzhou University, Fuzhou 350002, China;
d Marine College, Shandong University, Weihai 264209, China;
e Department of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia;
f State Key Lab of Transducer Technology, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China
The hydrogels domain was extended very fast owing to their capability to form multi-components network and by creating products with controlled properties for different applications [1]. Most polymer hydrogels are not self-healing and cannot be automatically repaired after damaged, because they are constructed by non-dynamic covalent bonds [2, 3]. By designing dynamic covalent bonds can endow polymer hydrogels with intrinsic self-healing capability in response to external stimuli (pH, light or temperature) [3-6]. Several approaches have been exploited, such as hydrogen bonds (H-bonds) [7], hydrophobic [8, 9], imine bonds [10], Diels-Alder click reaction and acylhydrazone bonds [11, 12], host-guest [13] or specific peptide interactions [14] to prepare self-healing materials. However, most of these polymer hydrogels with dynamic covalent reversible bonds were inherently vulnerable with low strength, and thereby are seldom studied in bone tissue regeneration and repair which requires new methods to prepare advanced hydrogels with strong mechanical robustness, adhesion and good osteoinduction.
Natural nanocomposites such as nacre and bone possess exceptional strength and toughness due to the effective regulatory mechanism and the electrostatic interactions between organic matrix (e.g., collagen) and inorganic species (e.g., hydroxyapatites (HAP), calcium carbonate) [15-18]. Inspired by the bionic-innovation strategy of natural nanocomposites, lots of work has been done to design hybrid hydrogels for bone tissue engineering and repair by using various polymer hydrogel-bioactive inorganic nanoparticle pair (Fe3O4-CS/telechelic difunctional poly(ethylene glycol) (DF-PEG) [19], bisphosphonated hyaluronan (HA-BP)/CaP NPs [20], BP- functionalized gelatin/bioactive glass particles [21], silk fibroin-based HA-BP·CaP [22], HA-BP/MgSiO3 NPs [23], alginate (Alg)/polyacrylamide (PAM)/mesoporous silica microrods (SBA-15) [24], CaP bone cements/glycidyl methacrylate derivatized dextran (Dex-MA) [25], collagen/HAP/double-network (DN) [26], HAP/swim bladder collagen/PDMAAm [27]). These nanocomposites have found to have excellent bone biocompatibility but weak strength [19, 21-23], good compressive strength but a weak osteoinduction ability [24, 25], relatively low flowability and diffusion in the donor site [20, 26, 27], respectively. Therefore, new strategies and methods are highly desired for the development of novel organic-inorganic hybrid hydrogels that possess excellent mechanical strength, controllable flowability and good osteoinductivity for bone regeneration and repair.
Recently, biocompatible chitosan (CS)-based hydrogel has shown great potential widely used in biomedicine since it contains dynamic covalent imine bond which is formed by the reversible Schiff base condensation reaction [19, 28]. The natural CS is cheap and biocompatible, but has relatively poor water solubility and weak chelation effect of amino group [29]. Few biocompatible materials have been designed by employing glycol chitosan (GCS) matrices, a typical derivative of CS, to strongly bind multivalent cations such as calcium ions (Ca2+) due to their rich −OH and −NH2 groups via metal ions-chelating mechanism [30-32]. Sodium alginate (SA) is abundant in ocean and has good biodegradability and biocompatibility, which can be easily oxidized into oxidized sodium alginate (OSA) by transforming adjacent −OH into −CH=O [33]. Chen and co-worker prepared a OSA-based hydrogel through the condensation reaction of the −CH=O groups of OSA with the −NH2 groups of N-carboxyethyl chitosan [34]. However, in practical bone regeneration, the above-mentioned hydrogels suffer the drawback of low compress strength and thus cannot support the bone defect. Calcium phosphate nanoparticles (CaP NPs) have broad applications in osteoporosis and fractures for its outstanding bioactivity and osteoinduction [35, 36]. It can release Ca2+ in hybrid biomaterials and simulate the native extracellular matrix (ECM). Chelation can occur between −OH, −NH2 of GCS and Ca2+ of CaP NPs [37]. In our previous study, bioactive HAP NPs can be crystallized in CS chains, but the incorporation of CaP NPs into self-healing polymer network for the osteoconduction of encapsulated stem cells (ESC) remains unexplored [38, 39]. Hence, the mechanical strength of the CS-based hybrid biomaterials can be enhanced along with the increased intermolecular interactions and viscoelasticity.
In this study, a novel kind of injectable CS-based biodegradable and self-healing hybrid (GCS-OSA-CaP) hydrogel, denoted as SHH-0.6, was designed and synthesized via a rapid (within 30 s), facile and easy gelation approach through exploiting the reversible Schiff-base reaction between OSA with −CH=O groups and GCS with −NH2 groups in the presence of CaP NPs (Scheme 1a). In the fabrication system of SHH-0.6 hydrogels, the molar ratio (R = M-NH2:M-CHO) of −NH2 from GCS and −CHO from OSA in hydrogel was 0.6, and the total concentration of CaP NPs was 6 wt%. By changing the R, a series self-healing hybrid hydrogels were obtained and denoted as SHH-R (R = 0.1, 0.3, 0.6, 0.9), and the relative dosage of CaP NPs in each SHH-R was maintained at 6 wt%. In practical bone regeneration, the hybrid hydrogel can be on-demand prepared because OSA and CaP NPs as raw materials can be readily synthesized in large quantity for long-term storage via a simple, one-time synthesis progress. The resulting SHH-0.6 hydrogel shows excellent reversible fluid-solid transformation in response to pH changes, rapid self-healing property and good injectability under physiological conditions without any external stimulus, and controlled drug release using rhodamine B as a model. The self-healing ability is ascribed to the dynamic equilibrium of the −CH=N− bonds in 3D hydrogel networks and homogeneous dispersion of CaP NPs (Scheme 1b). The mechanical strength of the hybrid SHH-0.6 hydrogel is significantly enhanced via the chelation between −OH, −NH2 of GCS and Ca2+ in the surface of CaP NPs and intermolecular H-bonds between GCS and CaP NPs. CaP NPs act as stable inorganic crosslinkers to stabilize the hydrogel network and hence improve the drug loading capacity and controlled release behavior. The hybrid SHH-0.6 hydrogel also exhibits superior degradability and biocompatibility as well as osteoinduction of ESC in vitro, opening up the possible applications in bone regeneration medicine, drug delivery or cell encapsulation as an injectable cytoskeleton.

|
Download:
|
| Scheme 1. (a) Chemical structures and reaction mechanism of the CGS-OSA-CaP hydrogel obtained by condensation reaction of the −CHO (from OSA) with the −NH2 (from GCS) mixed with CaP NPs, resulting in dynamic −CH=N− groups. (b) Schematic illustration of the self-crosslinking and self-healing process of GCS-OSA-CaP hydrogel. | |
{kind=link}
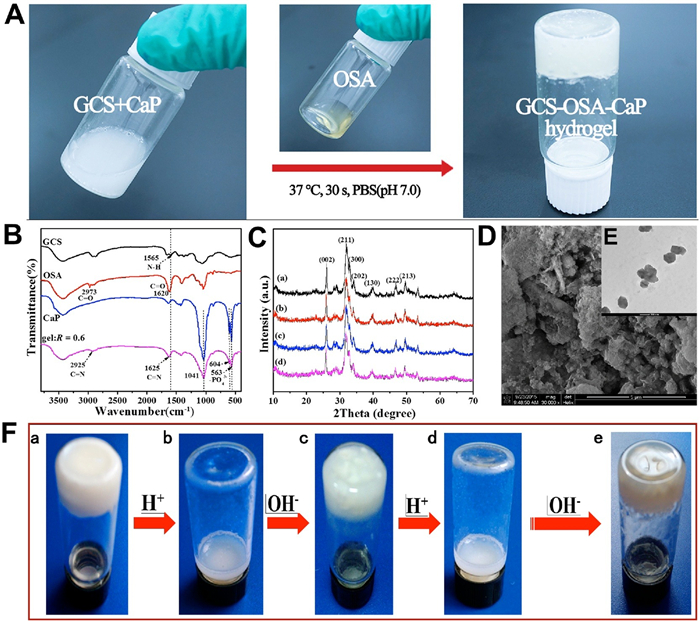
The whole gelation preparation approach is rapid and efficient, which only involves 30 s' vortex and then 90 s' standing at 37 ℃ without special equipment or environmental condition (Fig. 1A and Movie S1 in Supporting information). The hybrid hydrogel can be prepared on demand because OSA and CaP NPs as raw materials can be readily synthesized in large quantity for long-term storage. Fourier transfer infrared spectra of the composite before and after hydrogel formation were studied (Fig. 1B). The characteristic peaks at 2973 and 1620 cm−1 confirm the structure of OSA [40]. Compared to the spectrum of GCS and OSA, new peaks emerged at 2925 and 1625 cm−1 in the spectrum of the obtained SHH-0.6 hydrogel (Fig. 1B), and the characteristic absorption band of GCS at 1565 cm−1 (amide Ⅱ) disappeared in spectra of hydrogel [41, 42]. These results clearly indicate the formation of the Schiff base between GCS and OSA, namely, the –NH2 groups of GCS were crosslinked with −CH=O of OSA, generating −CH=N− bonds. The absorption peaks at 1041, 604 and 563 cm−1 are attributed to PO43- of CaP NPs (Fig. S1 in Supporting information) [43]. X-ray diffraction measurement (Fig. 1C) shows the main diffraction peaks at (211), (002), (300), (222) and (213) assigned to CaP NPs with a high crystallinity [44]. As R increased (R = M-NH2:M-CHO(0 ≤ R ≤ 1)), the high molecular weight GCS (Mw = 150, 000 Da) concentration increased, but the 6 wt% total concentration of the hydrogel remained the same, so the CaP quality of the hydrogel enhanced correspondingly. Thermogravimetric analysis (TGA) results show that the thermal stability of hydrogel gradually increased (Fig. S2 in Supporting information) as the R value increased from 0.1 to 0.9 mainly due to the increased crosslinking density (namely, more imine linkage formed). The CaP NPs exhibit cube morphology with uniform crystallite size of ~130 nm (Fig. 1E). The hybrid hydrogel (e.g., SHH-0.6) showed a porous and continuous 3D polymer network structure and homogeneous distribution of CaP NPs in accordance with the results of FT-IR and XRD (Fig. 1D, Figs. S3 and S4 in Supporting information).

|
Download:
|
| Fig. 1. Fabrication and characterization of GCS-OSA-CaP hydrogel. (A) Photographs demonstrate the gel formed between GCS-CaP and OSA in PBS (pH 7.0). (B) FT-IR spectra of GCS, OSA, CaP NPs, SHH-0.6. (C) XRD patterns of hydrogel (a) SHH-0.1, (b) SHH-0.3, (c) SHH-0.6, (d) SHH-0.9. (D) SEM images of SHH-0.6. Scale bar = 1 μm. (E) TEM images of CaP NPs. Scale bar = 200 nm. (F) The pH sensitivity of SHH-0.6: (a) origin hydrogel; (b) hydrogel decomposition after adding HCl (60 μL, 6 mol/L); (c) regeneration of the hydrogel after adding NaOH (60 μL, 6 mol/L); (d) decomposition of regenerated hydrogel after adding HCl (60 μL, 6 mol/L) and (e) regenerated hydrogel after six cycles. | |
{kind=link}
In the fabrication process of SHH-0.6 hydrogel, the pH value of the system has a crucial effect on the equilibrium of the imine linkage. SHH-0.6 gelatinized when it was soaked in PBS (pH 7.0), while the 3D network of SHH-0.6 collapsed and gradually liquefaction at pH 5.0. The pH stimuli-responsiveness of SHH-0.6 was indicated by liquefaction and gelatinization, re-liquefaction and re-gelatinization, repeated six times at least (Fig. 1F).
To study the self-healing behavior and mechanical strength of the SHH-0.6 hydrogel, rheological recovery tests were conducted. Hydrogel without CaP NPs was set as control (Figs. S5 and S6 in Supporting information). The results indicated that storage moduli (G') are higher than loss moduli (G") operating under different alternating quantity of frequency, time and strain (Figs. S7A–C in Supporting information). The increase of time and frequency causes little change in G' and G". As R varied from 0 to 0.6, the overall contents of imine linkage significantly increased and the chelation of CaP NPs as organic cross-linker was dramatically enhanced, resulting in an increased of G' from 717 ± 69 Pa to 3304 ± 237 Pa. While R further increases from 0.6 to 0.9, G' dropped to 2371 ± 237 Pa (Fig. S7D in Supporting information). Moreover, the photographs of the hydrogels with different mole ratio were intuitively shown in Fig. S8 (Supporting information). When the molar ratio was lower (e.g., R = 0.1), the precursor solution was not gelatinized. When the molar ratio becomes higher (e.g., R = 0.9), the hydrogel has no longer self-healing capability. So, the hybrid hydrogels (R = 0.6) were acted as the optimization for further in vitro tests.
As a result of the presence of rich dynamic −CH=N− bonds, the CaP-stabilized hydrogels exhibit macroscopic self-healing behavior (Fig. S9 and Video S2 in Supporting information). Two pieces of SHH-0.6 hydrogel cubes dyed in red and blue by Rhodamine B (RhB) and methylene blue (MB), respectively. They were individually separated into 27 small pieces (subcubes) of similar size (Figs. S9a and b). These small SHH-0.6 subcubes were remodelled to form a Rubik's cube (Fig. S9c). The boundaries between different pieces becomes blurred after standing for 6 h (Fig. S9d). Furthermore, no fissures were visible in the healed hydrogel Rubik's cube and the RhB and MB as the two model molecule drugs can rapidly diffuse within the hydrogels during soaking in PBS (pH 7.0) for 6–24 h (Figs. S9e–g).
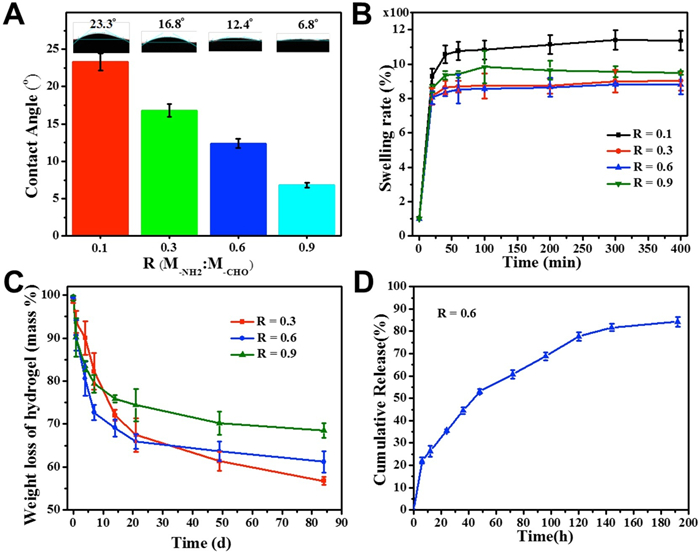
Physicochemical properties in vitro of the nanocomposite hydrogels were examined, including the contact angle, swelling property, degradability and drug release behavior (Fig. 2). With R increases, the water contact angle of the hydrogel decreases from 23.3° to 6.8° (Fig. 2A), indicating the decrease of crosslinking degree of polymer chains due to the interference effect of CaP NPs-GCS chelation. The SHH-0.6 hydrogel displayed a moderate swelling property inflated to 850% (Fig. 2B) and a suitable degradation ratio that gradually decreased to 61.20% ± 0.50% after 84 d of incubation (Fig. 2C and Fig. S10 in Supporting information). Moderate swelling property can promote the good binding force between the surrounding tissue and the hybrid hydrogel, which can effectively avoid the migration of the hydrogel as a filling material, and is also conducive to the controlled release of drug. Previous studies have demonstrated that Alendronate (ALN), the third generation of aminobisphosphonates, functioned as effective inhibitor of bone resorption by virtue of the high affinity with solid calcium phosphate in bone matrix and stability, are the first choice for the treatment of bone metabolic diseases such as osteoporosis. Based on this, the SHH-0.6 hydrogel loaded with ALN as a drug delivery nanosystem was designedly developed, which exhibited a sudden release of ALN at the beginning and the release rate decreased gradually with a cumulative release of 84.27% ± 0.50% (Fig. 2D and Fig. S11 in Supporting information). The sudden release (22.08% ± 0.50%) occurs in the first six hours due to the diffusion of cargo. It is possible that the cumulative release rate is a little higher for osteoinduction. However, the hybrid hydrogel could be prepared on demand. Herein, we could multi-inject drugs-loaded SHH-0.6 hydrogels to lesions at regular intervals during the healing time because of its excellent injectable, self-healing and easy-prepared ability.

|
Download:
|
| Fig. 2. (A) Contact angle, (B) swelling ratio and (C) degradation ratio of SHH-R (R=0.1, 0.3, 0.6, 0.9). (D) Cumulative release curves of SHH-0.6-ALN (0.01 mol/L PBS, pH 7.0, 37 ℃). Error ranges are standard deviations over n=3 samples. | |
{kind=link}
The injectability of hybrid SHH-0.6 was testified by two different colored SHH-0.6 cubes (Fig. S12a in Supporting information). They were injected into three different shaped molds by two 2 mL syringes and can be compressed into different shape on demand, such as quincunx, pentagram and heart shape (Figs. S12b and c in Supporting information). Two SHH-0.6 Rubik's cubes were reintegrated into three new different shaped monolithic hydrogels after 30 min self-healing (Fig. S12d in Supporting information), which still maintained their integrity after immersing in PBS (pH 7.0, 37 ℃) for 6–24 h (Figs. S12e–g in Supporting information), confirming an excellent self-healing performance.
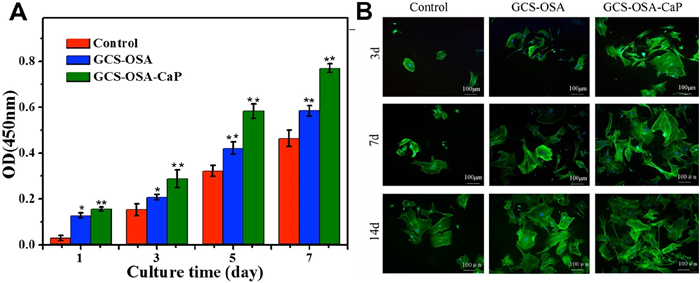
According to CCK-8 assay (Fig. 3A) and MTT assay (Fig. S13 in Supporting information), the cell viability of SHH-0.6 was more markedable with time continuing, in comparison to control group (*P < 0.05; **P < 0.01). The SHH-0.6 could significantly promote BMSCs attachment and proliferation. Furthermore, confocal images of BMSCs co-cultured with SHH-0.6 hydrogel for 3, 7 and 14 days exhibited normal cell morphology and abundant cell numbers (Fig. 3B). The live/dead staining presented less dead cells in SHH-0.6 hydrogel group (Fig. S14 in Supporting information).

|
Download:
|
| Fig. 3. (A) Cell toxicity evaluation by CCK8 assay after 1, 3, 5, 7 days' culture. Proliferation of BMSCs co-cultured with GCS-OSA hydrogel and SHH-0.6 under the same culture condition. Density of BMSCs was 1×105 cells/mL. Error ranges are standard deviations over n=3 samples. (B) Confocal images of BMSCs cultured on control, GCS-OSA hydrogel and SHH-0.6 for 3, 7 and 14 days. Mean values±standard deviation. (*P < 0.05; **P < 0.01) measured by one-way analysis of variance (ANOVA). | |
{kind=link}
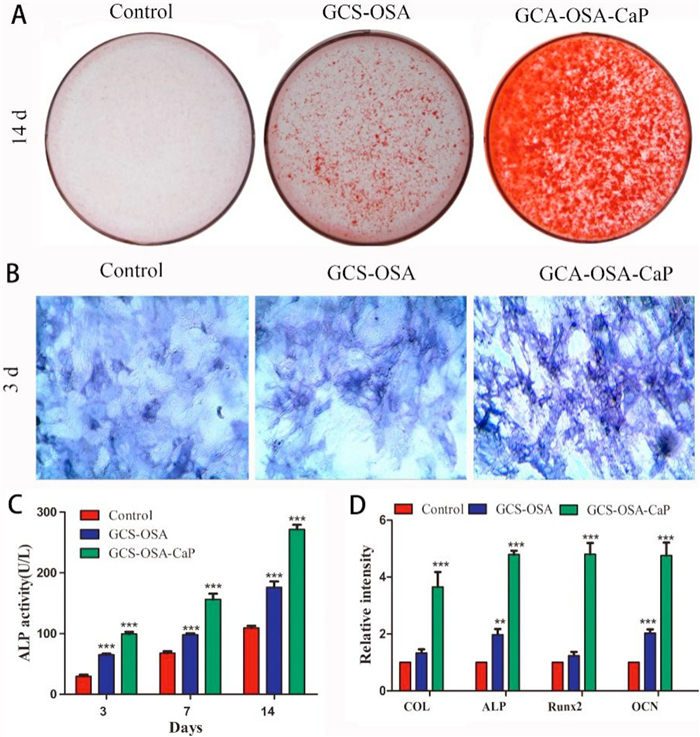
When BMSCs were co-cultured with hydrogels for 14 d, the calcium deposition was stained with Alizarin red (Fig. 4A). More visible calcium nodules were observed on SHH-0.6 compared to the GCS-OSA hydrogel and control group, suggesting that the SHH-0.6 hydrogel could promote matrix mineralization during BMSCs osteogenic differentiation. Moreover, fluorescence imaging reveals that BMSCs were distributed more sporadically in control group than in GCS-OSA and SHH-0.6 (Fig. 4B). Furthermore, ALP activity in SHH-0.6 hydrogel was higher than that in control group on 3, 7, 14 d (***P < 0.001) (Fig. 4C). Osteogenesis-related mRNA expressions were assessed by RT- PCR. As shown in Fig. 4D, COL, ALP, Runx2 and OCN mRNA in SHH-0.6 hydrogel are 156.4%, 142.7%, 288.9% and 134.7%, higher than those of the GCS-OSA hydrogels, demonstrating the important role of CaP NPs.

|
Download:
|
| Fig. 4. (A) Alizarin staining of calcium deposition by BMSCs in control, GCS-OSA hydrogel and SHH-0.6 group after 14 d of induction. (B) ALP staining after 3 d of induction. (C) Detection of alkaline phosphatase activity in BMSCs after 3, 7, 14 d. (D) Analysis of mRNA expression of the bone associated markers Col, ALP, RunX2 and OCN with qRT-PCR after 21 d of culture. (*P < 0.05, **P < 0.01, ***P < 0.001) measured by one-way analysis of variance (ANOVA). | |
{kind=link}
It is found in this study that the adding amount of CaP NPs can regulate the property of the hybrid hydrogel. Lower content of CaP NPs results in hydrogels with weak osteoinductivity, while too high CaP content gives rise to hydrogels poor self-healing performance. After optimizing the synthesis parameters, an appropriate amount of 6 wt% CaP NPs was introduced for the synthesis of hybrid hydrogels (denoted as SHH-0.6) with different molar ratio (R) of −CHO groups from OSA and −NH2 groups from GCS for further study. In this resulting hybrid hydrogel, CaP NPs probably function as an inorganic cross-linker to stabilize the hydrogel network and make successful conjugation to GCS. Such a pH response is due to the fact that Schiff base reaction is relatively stable under neuter and alkaline condition and normally sensitive to hydrolysis under acidic condition.
Moreover, the increased amount of CaP NPs-polymers and the enhanced interactions between them play a dominant role at the outset, while the unstable hydrogel networks show advantage at later stage. The results of macroscopic self-healing behavior reveals that the dynamic −CH=N− bonds in the hydrogel networks but not simple adhesions are vital in self-healing behavior in the interfaces of damaged SHH-0.6 hydrogels. Moderate swelling and hydrophilicity can produce good adhesion between the surrounding tissue and hydrogels, and then significantly restrains the movement of hydrogel network and the quick release of RhB. When the hydrogel reached the equilibrium swelling state, small drug molecules can overcome the constraints of the 3D hydrogel network and the electrostatic interactions of molecules. Moreover, the enhencement of swelling increases the contact area between the enzyme and hydrogel surface, accelerating the degradation rate of hydrogel. The minor difference between these physicochemical curves indicated that CaP concentration content exert almost negligible influences on drug release behavior. Among the obtained hydrogels, SHH-0.6 is more favorable for ESC due to the strong chelation interactions between GCS and CaP NPs and intermolecular H-bonds.
The drug-loaded or cell-seeded nanocomposite SHH-0.6 displays good injectability and remoldability after gelation, and thus are particularly suitable to different shaped bone lesions for defect filling, drug sustained release in bone engineering regeneration [34]. CCK-8 assay and MTT assay results indicates that the SHH-0.6 hydrogel is biologically friendly with good cytocompatibility to BMSCs/osteoblasts. The BMSCs exhibited clear spindle shape with more filopodia extensions on SHH-0.6 hydrogel compared with GCS-OSA and control group, implying that SHH-0.6 hydrogel can promote BMSCs adhesion and proliferation due to the presence of CaP NPs. The introduced CaP NPs can provide supporting effect for BMSCs spreading and cytoskeletal stretching, and they serve as a 'nutrition pool' for calcium nodule formation, thus promoting the BMSCs osteogenesis. Based on all these results, it can be concluded that the hybrid SHH-0.6 hydrogels show strong osteoinduction ability, which have promising orthopedic utility in bone tissue regeneration or drug delivery as an injectable scaffold.
An injectable CS-based self-healing hybrid hydrogel was on-demand designed via an easy, facile and rapid (within 30 s) gelation approach by the reversible Schiff-base condensation between −CH=O from OSA and −NH2 from GCS mixed with CaP NPs. Its raw materials can be mass prepared in advance via a simple and efficient synthesis process. The nanocomposite hydrogel exhibits excellent properties of sensitive pH-response, facile injectability and strong self-healing capability, as well as sustained drug release. The robust dynamic interaction of the reversible imine bonds in 3D hydrogel networks confers self-healing ability. Most importantly, the hybrid hydrogel displays excellent biodegradable, biocompatibility and efficient osteoinduction, and significantly promotes BMSCs osteogenic differentiation in vitro. The introduction of CaP NPs to hydrogel network supports BMSCs spreading and cytoskeletal stretch, and thereby accelerates the osteogenesis by encapsulated BMSCs. CaP NPs could provide Ca2+ for BMSCs to form calcium nodules. Based on the above results, a novel approach to establish hybrid hydrogel is provided for potential orthopedic applications such as bone defect filling, drug delivery or cell transplantation as an injectable drug-loaded hydrogel scaffold.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Key Research and Development Program of China (No. 2017YFC1104102), National Natural Science Foundation of China (Nos. 31370958, 21875044) and Key Program of Natural Science Foundation of Fujian Province (No. 2018Y0056). The authors extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP-17-94(2).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.001.
| [1] |
M. T. Nistor, A.P. Chiriac, L.E. Nita, C. Vasile, L. Verestiuc, Compos. Part. B -Eng. 43 (2012) 1508-1515. DOI:10.1016/j.compositesb.2011.10.015 |
| [2] |
D. Mawad, A. Artzyschnirman, J. Tonkin, et al., Chem. Mater. 28 (2016) 6080. DOI:10.1021/acs.chemmater.6b01298 |
| [3] |
S. Pina, J.M. Oliveira, R.L. Reis, Adv. Mater. 27 (2015) 1143-1169. DOI:10.1002/adma.201403354 |
| [4] |
N.K. Singh, D.S. Lee, J. Control. Release 193 (2014) 214-227. DOI:10.1016/j.jconrel.2014.04.056 |
| [5] |
A. Alam, Q. Meng, G. Shi, et al., Compos. Sci. Technol. 127 (2016) 119-126. DOI:10.1016/j.compscitech.2016.02.024 |
| [6] |
H. Qin, T. Zhang, N. Li, H.P. Cong, S.H. Yu, Nat. Commun. 10 (2019) 2202. DOI:10.1038/s41467-019-10243-8 |
| [7] |
J. Cui, C.A. Del, Chem. Commun. 48 (2012) 9302-9304. DOI:10.1039/c2cc34701f |
| [8] |
A. Phadke, C. Zhang, B. Arman, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 4383. DOI:10.1073/pnas.1201122109 |
| [9] |
Z. Lu, S. Liu, Y. Le, et al., Biomaterials 218 (2019) 119190. DOI:10.1016/j.biomaterials.2019.05.001 |
| [10] |
Y. Zhang, L. Tao, S. Li, Y. Wei, Biomacromolecules 12 (2011) 2894-2901. DOI:10.1021/bm200423f |
| [11] |
F. Yu, X. Cao, J. Du, G. Wang, X. Chen, ACS Appl. Mater. Interfaces 7 (2015) 24023. DOI:10.1021/acsami.5b06896 |
| [12] |
X. Ji, Z. Li, X. Liu, et al., Adv. Mater. 31 (2019) e1902365. DOI:10.1002/adma.201902365 |
| [13] |
M. Nakahata, Y. Takashima, H. Yamaguchi, A. Harada, Nat. Commun. 2 (2011) 511. DOI:10.1038/ncomms1521 |
| [14] |
C. Tondera, R. Wieduwild, E. Röder, et al., Adv. Funct. Mater. 27 (2017) 1605189. DOI:10.1002/adfm.201605189 |
| [15] |
Y. Chen, Y. Xianyu, X. Jiang, Acc. Chem. Res. 50 (2017) 310. DOI:10.1021/acs.accounts.6b00506 |
| [16] |
A. Gautieri, S. Vesentini, A. Redaelli, M.J. Buehler, Nano Lett. 11 (2011) 757-766. DOI:10.1021/nl103943u |
| [17] |
J. Lu, C. Cheng, Y.S. He, et al., Adv. Mater. 28 (2016) 4025-4031. DOI:10.1002/adma.201505375 |
| [18] |
S.J. Wang, D. Jiang, Z.Z. Zhang, et al., Adv. Mater. 31 (2019) 1904341. DOI:10.1002/adma.201904341 |
| [19] |
H. Petite, V. Viateau, W. Bensaïd, et al., Nat. Biotechnol. 18 (2000) 959-963. DOI:10.1038/79449 |
| [20] |
M.R. Nejadnik, X. Yang, M. Bongio, et al., Biomaterials 35 (2014) 6918. DOI:10.1016/j.biomaterials.2014.05.003 |
| [21] |
M. Diba, W.A. Camargo, M. Brindisi, et al., Adv. Funct. Mater. 27 (2017) 1703438. DOI:10.1002/adfm.201703438 |
| [22] |
L. Shi, F. Wang, W. Zhu, et al., Adv. Funct. Mater. 27 (2017) 1700591. DOI:10.1002/adfm.201700591 |
| [23] |
L.A.C. Diaz, A. Saiani, J.E. Gough, A.F. Miller, J. Tissue Eng. 5 (2014) 1-12. |
| [24] |
S. Choi, Y.J. Choi, M.S. Jang, et al., Adv. Funct. Mater. 27 (2017) 1703826. DOI:10.1002/adfm.201703826 |
| [25] |
P. Xu, J. Huang, P. Cebe, D.L. Kaplan, Biomacromolecules 9 (2008) 1551-1557. DOI:10.1021/bm701365x |
| [26] |
T. Nonoyama, S. Wada, R. Kiyama, et al., Adv. Mater. 28 (2016) 6740-6745. DOI:10.1002/adma.201601030 |
| [27] |
K. Zhang, Q. Feng, J. Xu, et al., Adv. Funct. Mater. 27 (2017) 1701642. DOI:10.1002/adfm.201701642 |
| [28] |
B. Ye, S. Zhang, R. Li, L. Li, L. Lu, C. Zhou, Compos. Sci. Technol. 156 (2018) 238-246. DOI:10.1016/j.compscitech.2017.12.032 |
| [29] |
N. Bhattarai, J. Gunn, M. Zhang, Adv. Drug Deliv. Rev. 62 (2010) 83-99. DOI:10.1016/j.addr.2009.07.019 |
| [30] |
J. Feng, Y. Chen, F. Li, et al., Molecules 22 (2017) 1253. DOI:10.3390/molecules22081253 |
| [31] |
B. Erdinc, R.J. Neufeld, Drug Dev. Ind. Pharm. 37 (2011) 619-627. DOI:10.3109/03639045.2010.533681 |
| [32] |
Z.K. Cui, S. Kim, J.J. Baljon, et al., Nat. Commun. 10 (2019) 3523. DOI:10.1038/s41467-019-11511-3 |
| [33] |
J. Zhang, Q. Wang, A. Wang, Acta Biomater. 6 (2010) 445. DOI:10.1016/j.actbio.2009.07.001 |
| [34] |
Z. Wei, J.H. Yang, Z.Q. Liu, et al., Adv. Funct. Mater. 25 (2015) 1352-1359. DOI:10.1002/adfm.201401502 |
| [35] |
J.A. Inzana, D. Olvera, S.M. Fuller, et al., Biomaterials 35 (2014) 4026. DOI:10.1016/j.biomaterials.2014.01.064 |
| [36] |
W. Li, J. Kang, Y. Yuan, et al., Compos. Sci. Technol. 128 (2016) 58-64. DOI:10.1016/j.compscitech.2016.03.013 |
| [37] |
L. Li, J. Li, J. Guo, et al., Adv. Funct. Mater. 29 (2018) 1807356. |
| [38] |
J. Chen, P. Pan, Y. Zhang, S. Zhong, Q. Zhang, Colloids Surf. B 134 (2015) 401-407. DOI:10.1016/j.colsurfb.2015.06.072 |
| [39] |
P. Pan, J. Chen, T. Fan, et al., Colloids Surf. B 140 (2015) 50-59. |
| [40] |
C. Gao, M. Liu, J. Chen, X. Zhang, Polym. Degrad. Stab. 94 (2009) 1405-1410. DOI:10.1016/j.polymdegradstab.2009.05.011 |
| [41] |
H. Zhang, S. Syed, Org. Lett. 12 (2010) 708-711. DOI:10.1021/ol902722y |
| [42] |
B. Yang, Y. Zhang, X. Zhang, et al., Polym. Chem. 3 (2012) 3235-3238. DOI:10.1039/c2py20627g |
| [43] |
E. Charrière, S. Terrazzoni, C. Pittet, et al., Biomaterials 22 (2017) 2937-2945. |
| [44] |
M.P. Ginebra, C. Canal, M. Espanol, D. Pastorino, E.B. Montufar, Adv. Drug Delivery Rev. 64 (2012) 1090-1110. DOI:10.1016/j.addr.2012.01.008 |