 2021, Vol. 32
2021, Vol. 32 


b State Key Lab of Transducer Technology, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China;
c Department of Chemistry, College of Science, King Saud University, PO Box 2455, Riyadh 11451, Saudi Arabia
Sulfuric acid (H2SO4) is an important catalyst for production of many important industrial chemicals, and more than 15 million tons of H2SO4 are consumed per year in industry. However, the used H2SO4 is hardly separated from the homogeneous reaction systems, resulting in massive emissions of H2SO4/sulfate and severe harm to environment as well. Therefore, it is urgently desired to explore strong solid acids with high catalytic activity and long-term stability as a substitute for the unrecyclable liquid acids [1]. For example, as a kind of typical solid acids, cation-exchanged resins (e.g., perfluorosulfonic acid) are widely used as proton conductors in polymer electrolyte fuel cells (PEFCs), which can improve the fuel efficiency and reduce the usage of noble-metal catalysts. However, the polymer-based solid acids are still less active, less stable and more expensive than liquid acids [2-8], limiting their practical applications in PEFCs and other catalytic reactions. Therefore, it is of great significance to design and synthesize low-cost stable inorganic or organic materials with enough strong protonic acidity for use as highly qualified and recyclable solid acid catalysts.
In recent years, carbon materials, such as carbon nanotubes, mesoporous carbons, carbon dots, graphene, have been extensively synthesized and applied in catalysis, environmental protection, renewable energy storage, etc., mainly due to their high stability, easy scalability and adjustable physical/chemical properties [9-14]. Through sulfonation and incomplete carbonization of polycyclic aromatic compounds (such as naphthalene) in concentrated H2SO4, Hara and co-workers prepared carbon-based solid acids with high density of sulfonic acid groups (SO3H). The SO3H-bearing carbon materials showed strong protonic acidity, and were utilized as superior solid acid catalysts for diverse acid-catalyzed reactions [15-21]. Besides naphthalene, other low-cost widespread hydrocarbons can also be used as the starting carbon sources, such as polycyclic aromatic hydrocarbons in coal tar or petroleum pitch (anthracene, perylene, coronene, etc.), natural organic matter (sugars, starch, cellulosic materials, etc.) [15, 17, 19, 20]. Recently, Yang et al. designed several solid acid catalysts based on hybrid hollow or core-shell nanospheres, which were composed of sulfonated polystyrene (PS-SO3H) as cores and mesoporous silica as shells [22-24]. The composite materials showed much higher activity and better recyclability than Amberlyst-15 and concentrated H2SO4 in acid-catalyzed reactions. The solid acid catalysts can be easily collected for reuse by centrifugation or filtration from the heterogeneous systems; however, it still remains a great challenge in practical reactions due to their low separation efficiency and high energy consumption.
During past several years magnetically recoverable catalysts have attracted tremendous interests, mainly because they possess a promising accessibility of facile separation and improved reusability from reaction systems under an external magnet [25-29]. For example, Tai et al. synthesized Fe3O4@SiO2@mSiO2 nanoparticles, which owned sulfonic acid-functionalized mesoporous silica shell and demonstrated improved activity and facile recovery in esterification [30]. Compared to amorphous silica, carbon materials can be easily and stably modified during sulfonation process. The carbon-based microspheres grafted with SO3H groups possess better chemical inertness, higher thermal stability and stronger acidity than the silica-based sulfonic acid materials [31]. Therefore, many strategies have been adopted for the synthesis of magnetic carbonaceous solid acid catalysts [32, 33]. To the best of our knowledge, until now little work has been done to rationally synthesize carbon-based magnetic solid acid catalysts with well-defined structure and functionality. Ideally, from the viewpoint of practical applications, these catalyst materials should possess uniform core-shell structure consisting of magnetic cores and SO3H-bearing incompletely-carbonized shells so as to provide surface affinity to reaction medium and obtain convenient recycling catalysts. In past decades, our group has developed a novel surfactant-directed interface co-assembly strategy to design magnetic core-shell (or yolk-shell) structured microspheres, which are usually composed of Fe3O4 inner cores and permeable mesoporous shells [34-40]. In order to improve the stability and tune the surface property of Fe3O4 particles, dense protective layers of nonporous resorcinol-formaldehyde (RF) resins were deposited on Fe3O4 cores in advance, which were beneficial for subsequent uniform assembly of mesoporous shells [34, 35, 41]. Because of the strong complexation between phenolic hydroxyl groups and iron ions, resorcinol molecules can be strongly attached onto the surface of Fe3O4 particles, and thus uniform RF resin shells are easily formed by controllable polymerization of resorcinol with formaldehyde [34]. The deposited RF resins also contain abundant benzene ring groups similar with the polycyclic aromatic hydrocarbons described above [15-21], which can be incompletely carbonized through high-temperature or sulfonation treatments [42, 43]. Herein, we first synthesized monodisperse magnetic core-shell Fe3O4@RF microspheres by controllable interface polymerization between resorcinol and formaldehyde on magnetic Fe3O4 particles. Then the microspheres were calcined in N2 atmosphere to form Fe3O4@carbon microspheres (Fe3O4@C) through in situ carbonization of RF shells under catalytic assistance of iron ions. After further surface sulfonation of the carbon shells, core-shell Fe3O4@C-SO3H microspheres with rich sulfonic acid on surface were obtained and utilized as the solid acid catalyst for acetalization reaction between benzaldehyde and ethylene glycol, showing superior catalytic activity, selectivity and recyclability.
All reagents in analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd. (China) and used as received, including absolute ethanol (C2H5OH), resorcinol (C6H4(OH)2), formaldehyde (HCHO, 37 wt%), concentrated ammonia solution (NH3·H2O, 28 wt%), fuming sulfuric acid (H2SO4·SO3, with 50% free SO3), benzaldehyde (C6H5CHO), ethylene glycol (C2H6O2), cyclohexane (C6H12) and so on. Deionized water was used where needed.
Synthesis of magnetic core-shell Fe3O4@RF and Fe3O4@C microspheres: Hydrophilic water-dispersible Fe3O4 particles with a diameter of ~ 90 nm were synthesized by a solvothermal method reported before [44]. The as-made Fe3O4 particles of 20 mg were firstly dispersed in a glass flask containing 32 mL of ethanol and 80 mL of water under ultrasonication treatment for 15 min. After mechanical stirring (300 rpm) at 30 ℃ for 5 min, 1 mL of concentrated ammonia solution and 0.40 g of resorcinol were added successively to the solution with further stirring for 10 min. Afterward, formaldehyde of 0.60 mL was added dropwise with a syringe and the solution was continuously stirred for another 10 h. The solid product was separated by a magnet, followed by washing with 95% ethanol solution three times and drying at 70 ℃ for 12 h, to obtain core-shell Fe3O4@RF (RF=resorcinol-formaldehyde) microspheres. The microspheres were then calcined in N2 atmosphere at different temperatures (300−800 ℃) for 4 h (heating rate =1 ℃/min), and a series of Fe3O4@C-x microspheres (x stands for the calcination temperature) with carbon shells of different carbonation degrees were obtained. In the further catalysis process, RF microspheres were also synthesized for comparison, which were not coated on Fe3O4 particles, and were further carbonized in N2 atmosphere at 600 ℃ (denoted as C-600).
Synthesis of Fe3O4@C-x-SO3H microspheres: 20 mg of Fe3O4@C-x microspheres were put into a small crucible, which was then placed in a Teflon-lined stainless-steel autoclave holding 7 mL fuming sulfuric acid in the bottom. After sulfonation treatment at 60 ℃ for 48 h, the sample was washed repeatedly in hot water of 80 ℃ to remove physically adsorbed H2SO4. The residual H2SO4 in eluent was detected by adding BaCl2 solution until no formation of BaSO4 precipitation. After separation by a magnet, Fe3O4@C-x-SO3H microspheres were dried at 70 ℃ for 12 h for further use. For comparison, the carbon microspheres were also sulfonated to form C-600-SO3H.
Acidity determination and acetalization reaction of Fe3O4@C-x-SO3H microspheres: The sample of Fe3O4@C-x-SO3H (20 mg) was added into 20 mL of NaCl solution (2 mol/L) with stirring at room temperature for 24 h to exchange H+ with Na+ ions [22-24]. After separating the Na+-exchanged microspheres (Fe3O4@C-x-SO3Na) by a magnet, the remained solution was accurately titrated with 6.5 mmol/L NaOH solution to calculate the acid amounts of Fe3O4@C-x-SO3H samples.
The acetalization reaction between benzaldehyde and ethylene glycol was conducted in a three-necked flask. The sample of Fe3O4@C-x-SO3H (20 mg) was firstly dispersed in 10 mL of cyclohexane under ultrasonic treatment for 10 min, and then 1.53 mL of benzaldehyde (15 mmol) was added in the flask under mechanical stirring. Before reaction the air in the flask was completely removed in advance by injecting pure N2. Ethylene glycol of 0.84 mL (15 mmol) was injected into the flask when reaction temperature rose to 90 ℃. During reaction the intermediate liquid products were separated and analyzed by gas chromatography-mass spectrometry (GC–MS). After reaction the used catalyst was separated by a magnet, and the repeat experiments was conducted to test the recyclability under the same conditions. For comparison, the total acid amount and catalytic activity of C-600-SO3H were also measured in the same procedure.
X-ray powder diffraction (XRD) was conducted on Bruker D4 X-ray powder diffractometer (Bruker, Germany) between 10° and 80°/2θ (scan rate=0.02°/min), using Cu Kα radiation under working voltage of 40 kV and current of 40 mA. Scanning electron microscopy (SEM) was carried out on S-4800 field-emission scanning electron microscope (Hitachi, Japan) under working voltage of 20 kV. Transmission electron microscopy (TEM) was conducted on JEOL-2011 transmission electron microscope (JEOL, Japan) under acceleration voltage of 200 kV. Fourier-transform infrared (FT-IR) spectra were recorded with a KBr method on Nicolet Fourier spectrometer (Thermo Fisher, USA). Raman spectra were recorded in anti-Stokes range of 500-2000 cm−1 on inVia Raman microscope (Renishaw, USA) equipped with He-Gd laser at excitation wavelength of 532 nm. The magnetic property was measured on a superconducting quantum interference device at 300K. The concentration of reagents and products in acetalization reaction was analyzed by GC–MS on GC-7900 gas chromatographer (Techcomp, USA).
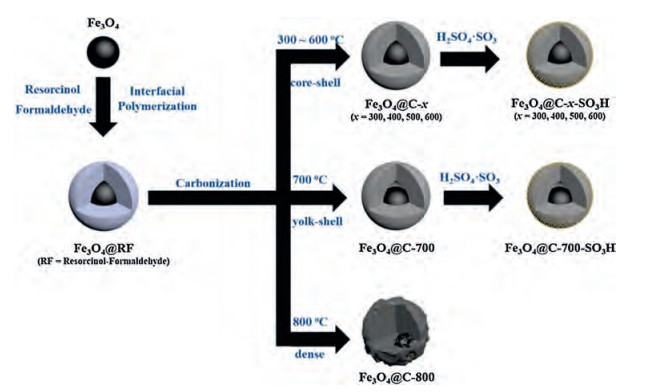
Scheme 1 illustrates the synthetic route of sulfonic acid-functionalized magnetic core-shell Fe 3O4@carbon (Fe3O4@C-x-SO3H) microspheres. First, a solvothermal method was used to synthesize Fe3O4 particles as magnetic cores. Second, through interfacial polymerization between resorcinol and formaldehyde under alkaline conditions, RF resin layers were uniformly deposited on the surface of Fe3O4 particles, resulting in magnetic core-shell Fe3O4@RF microspheres. Third, the microspheres were controllably calcined in N2 atmosphere at different temperatures. During this process the outer RF shells were partially carbonized into graphitic skeletons to form magnetic core-shell Fe3O4@C-x microspheres (x stands for the calcination temperature). Finally, through sulfonation treatment in fuming H2SO4, sulfonic acid groups (SO3H) were generated on the surface of carbon shells, and sulfonic acid-functionalized magnetic core-shell Fe3O4@C microspheres (Fe3O4@C-x-SO3H) were obtained.

|
Download:
|
| Scheme 1. Synthetic illustration of the sulfonic acid-functionalized magnetic core-shell Fe3O4@carbon microspheres through interfacial polymerization, carbonization in N2 atmosphere at different temperatures and following sulfonation treatment. | |
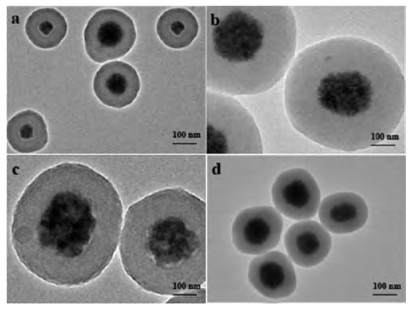
The magnetic Fe3O4 particles were synthesized by a simple solvothermal method [44], and TEM image (Fig. 1a) indicates that the as-synthesized Fe3O4 particles possessed uniform spherical morphology with an average diameter of ~ 90 nm, and were composed of a great number of tiny Fe3O4 nanocrystals of ~10 nm (Fig. 1b). Since many citrate groups were anchored on the rough surface, the polycrystalline Fe3O4 particles exhibited great hydrophilicity and high dispersibility in polar solvent such as ethanol aqueous solution. Moreover, resorcinol molecules were able to attach onto the surface of Fe3O4 due to strong complexation between iron ions and phenolic hydroxyl groups. Therefore, RF resin layers could be well deposited on the surface of Fe3O4 through stepwise polymerization of attached resorcinol with formaldehyde. In general, both hydrogen ions (H+) and hydroxide ions (OH−) are feasible catalysts by helping to link resorcinol with formaldehyde molecules, and H+ ions can help reduce gelation time in the sol-gel process, with a different polymerization mechanism against OH− ions [45]. However, acid solution can surely damage the structure of Fe3O4, so in this case, weak alkaline solution of ammonia (about pH 9) was selected as a buffer catalyst solution for polymerization between resorcinol and formaldehyde. The spherical morphology of Fe3O4 particles was still preserved after coating RF shells, and the formed Fe3O4@RF microspheres showed uniform core-shell structures and tunable shell thicknesses from 40 nm to 200 nm through changing the amounts of resorcinol and formaldehyde with a constant volume ratio of 1:1.5 (Figs. 1c–e). SEM image of the inorganic-organic hybrid Fe3O4@RF (Fig. S1 in Supporting information) displays many monodisperse microspheres with bright centers and dark surroundings, revealing uniform particle sizes and distinct core-shell structures. The shell thickness of RF layers plays a vital role in following carbonization process. Because thin shells (< 40 nm) can easily shrink and collapse during calcination, and thick shells (> 200 nm) can weaken magnetic property of the whole microspheres, herein, the Fe3O4@RF microspheres with an appropriate shell thickness of 90 nm were chosen for further carbonization and sulfonation.

|
Download:
|
| Fig. 1. TEM images of (a, b) Fe3O4 particles and Fe3O4@RF microspheres with shell thickness of (c) 40 nm, (d) 90 nm and (e) 200 nm, respectively. | |
Magnetic core-shell Fe3O4@C microspheres were obtained by carbonization of Fe3O4@RF with shell thickness of 90 nm under N2 atmosphere at different temperatures. TEM images indicate that the compact and core-shell structures were well maintained in the temperature range of 300−600 ℃ (Fig. 2), exhibiting monodisperse spherical morphology. The diameter of Fe3O4 particles and thickness of carbon shells in each sample were also quite close to those of the corresponding as-prepared Fe3O4@RF microspheres, and no obvious distortion or collapse was observed (Fig. 1d), revealing high thermal stability of the carbon shells between 300 ℃ and 600 ℃. Therefore, the optimal shell thickness of Fe3O4@C microspheres for further sulfonation is 90 nm. When carbonization temperature rose to 700 ℃, huge cavities appeared inside the carbon shells (Fig. S2a in Supporting information), resulting in typical yolk-shell structures. Notably, average size of the inner Fe3O4 cores increased to about 200 nm, but a slight deformation of spherical morphology was observed. At the same time the thickness of carbon shells greatly decreased to ~ 40 nm. These results imply that an intense reaction (e.g., carbothermal reduction) occurred between RF resins and Fe3O4 particles at 700 ℃, leading to formation of much denser carbon skeletons. When calcination temperature rose to 800 ℃, the core-shell structure of Fe3O4@RF microspheres completely collapsed, and only a few residues of Fe3O4 inside were observed (Fig. S2b in Supporting information). The particles of ~ 400 nm possess irregular spherical morphology and enclosed cavities, indicating a strong reaction between Fe3O4 and carbon shells at 800 ℃.

|
Download:
|
| Fig. 2. TEM images of Fe3O4@C microspheres obtained by carbonization of Fe3O4@RF microspheres with shell thickness of 90 nm at different temperatures (300-600 ℃): (a) Fe3O4@C-300, (b) Fe3O4@C-400, (c) Fe3O4@C-500, (d) Fe3O4@C-600. | |
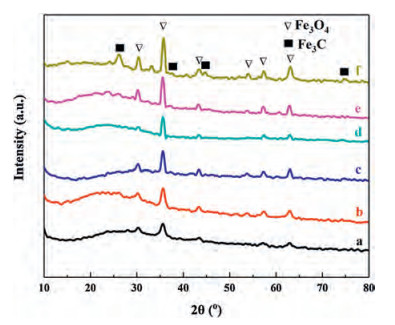
XRD pattern (Fig. 3) of Fe3O4@C microspheres displays six typical diffraction peaks, which were well indexed to the reflections of (220), (311), (400), (422), (511) and (440) lattice planes of Fe3O4 (JCPDS card No.19−0629), respectively, indicating existence of crystalline Fe3O4 under carbonization at the temperature ranging from 300 ℃ to 800 ℃. The intensity of main peak of Fe3O4 at around 35.4° increases as the temperature rises, indicating enlargement of the particle size of Fe3O4 nanocrystals due to their agglomeration and ripening at higher temperatures. Broad peaks in the range of 20°-30°/2θ were attributed to amorphous carbons in shells of Fe3O4@C microspheres, which were formed by carbonization of RF resins at high temperature. Besides of diffraction peaks about Fe3O4, both Fe3O4@C-700 and Fe3O4@C-800 show additional peaks assigned to (210), (211) and (112) lattice planes (JCPDS card No. 89-2867) of crystallized Fe3C (Fig. 3e and f), which was formed by deep reaction between RF resins and Fe3O4 particles when calcination temperature was over 700 ℃. The generation of Fe3C impurity in Fe3O4@C microspheres can greatly boost the appearance of yolk-shell structure in Fe3O4@C-700 and the collapse of core-shell structure of Fe3O4@C-800.

|
Download:
|
| Fig. 3. XRD patterns of Fe3O4@C microspheres obtained by carbonization of Fe3O4@RF microspheres with shell thickness of 90 nm at different temperatures (300-800 ℃): (a) Fe3O4@C-300, (b) Fe3O4@C-400, (c) Fe3O4@C-500, (d) Fe3O4@C-600, (e) Fe3O4@C-700 and (f) Fe3O4@C-800. | |
All the Fe3O4@C samples display two Raman bands (Fig. 4a) centered at 1365 and 1589 cm−1, which are assigned to D peak and G peak of graphitic carbon, respectively. Generally speaking, the intensity of D peak is associated with the structural disordering of amorphous or quasi-crystalline carbon materials, caused by regional edge modification of the plane structure due to lack of long-range order, while the G peak corresponds to the base surface E2g mode (stretching vibration) of the crystalline graphite. The intensity of G peak can indicate the ordering degree of surface graphene layer with sp2-hybridized carbon atoms, and the intensity ratio of D peak to G peak (ID/IG) is used to compare the graphitization degrees among different carbon materials. In general, smaller ID/IG value indicates higher regularities and ordering degrees of carbon [46]. The ID/IG value decreased as the carbonization temperature increased from 300 ℃ to 600 ℃ (Fig. 4b), revealing a gradual improvement of graphitization degree of carbon skeletons under catalytic assistance of iron ions. Among the six calcined samples, Fe3O4@C-600 possessed the smallest ID/IG value (0.99), indicating the highest graphitization degree of the carbon shells at 600 ℃. Although higher calcination temperature eventually resulted in enhanced carbon crystallization and improved graphitization degrees in the temperature range of 300 ~ 600 ℃, the graphitization degree was reduced when calcination temperature was over 600 ℃, mainly due to the formation of Fe3C impurity by excessive reaction between RF resins and Fe3O4 particles. The result was quite consistent with that of XRD patterns (Fig. 3), in which Fe3C was observed in Fe3O4@C-700 and Fe3O4@C-800. Therefore, the carbonization temperature of 600 ℃ was the most suitable for Fe3O4@RF graphitization to obtain qualified Fe3O4@C microspheres with intact and highly-graphitic carbon shells.

|
Download:
|
| Fig. 4. (a) Raman spectra and (b) the corresponding ID/IG values of Fe3O4@C microspheres obtained by carbonization of Fe3O4@RF microspheres with shell thickness of 90 nm at different temperatures (300-800 ℃): (a) from bottom to top, (b) from left to right: Fe3O4@C-300, Fe3O4@C-400, Fe3O4@C-500, Fe3O4@C-600, Fe3O4@C-700 and Fe3O4@C-800. | |
During the sulfonation process, sulfonic acid groups (SO3H) were chemically bonded onto the surface of carbonic layers of Fe3O4@C microspheres with a shell thickness of 90 nm in fuming H2SO4 vapor. It's well known that both concentrated H2SO4 aqueous solution and fuming H2SO4 are used as the sulfonation agents. However, direct exposure of Fe3O4@C microspheres in acid solution may seriously destroy the magnetic Fe3O4 cores. In this work the indirect exposure by fuming H2SO4 vapor can well preserve the stabilities of Fe3O4@C microspheres, because much less H+ ions are accessible to the microspheres. As shown in FT-IR spectra (Fig. S3 in Supporting information), each sample of Fe3O4@C-x-SO3H (x=300–700) exhibits an apparent absorption peak centered at about 1054 cm−1, which is attributed to the symmetric stretching vibration of S=O bonds. Moreover, some other peaks at 581, 1167, 1377 and 1415 cm−1 can also be observed, confirming successful graft of SO3H groups on Fe3O4@C microspheres [20, 22-24]. The broad peak at about 1625 cm−1 can be assigned to carbon-carbon bonds in graphite, suggesting partial carbonization of RF layers catalyzed by iron ions. In addition, no absorption bands about Fe3O4 are observed, indicating that Fe3O4 cores were well covered by carbon shells even after high-temperature carbonization and sulfonation treatment of the core-shell microspheres. Moreover, the magnetic property of the materials was characterized by using a superconducting quantum interference device (Fig. S4 in Supporting information). The Fe3O4@C-600-SO3H microspheres showed lower magnetization saturation value (10.6 emu/g) than Fe3O4 particles (59.9 emu/g), mainly because coating of RF layers onto Fe3O4 and followed carbonization treatment at 600 ℃ could inevitably reduce the magnetization saturation value. Even so, the Fe3O4@C-600-SO3H microspheres could be rapidly and completely separated from liquid under an external magnetic field. Besides, both samples showed no remanence with a great superparamagnetic ability, which stemmed from the intrinsic characteristics of magnetite nanocrystals in Fe3O4 particles [26]. Therefore, the core-shell Fe3O4@C-x-SO3H microspheres with both sulfonic acid groups and magnetic properties own a promising potential to be used as the magnetically recyclable solid acid catalysts, which are easily separated and reused for next-cycle evaluation.
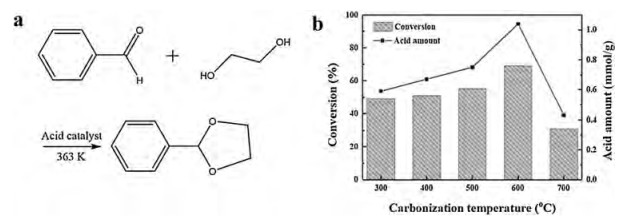
Acid amounts of the magnetic catalysts were determined by a titration method (Table 1), which increased from 0.59 mmol/g to 1.04 mmol/g at the carbonization temperature ranging from 300 ℃ to 600 ℃. The sequence shows a good relationship with the graphitization degrees of Fe3O4@C microspheres (Fig. 4b), demonstrating feasible sulfonation and stable graft of SO3H groups on graphitic carbon shells [47]. Although Fe3O4@C-700 showed comparable graphitization degree with Fe3O4@C-600, Fe3O4@C-700-SO3H exhibited the lowest acid amount (0.43 mmol/g). Therefore, the number of acid sites was greatly reduced by decrease of grafted SO3H groups on the surface of carbon shells due to formation of Fe3C impurity at the temperature higher than 700 ℃.
|
|
Table 1 Total acid amounts of different Fe3O4@C-x-SO3H, C-600-SO3H and Fe3O4@C-600 materials and the conversions of benzaldehyde in the catalyzed acetalization reactions. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The catalytic performance of the Fe3O4@C-x-SO3H catalysts was tested for the acetalization reaction between benzaldehyde and ethylene glycol (Fig. 5a), which is a typical acid-catalyzed reaction for protecting carbonyl groups in multi-procedure organic synthesis. During the catalysis process, the organic components in reaction solution were extracted and analyzed by GC–MS to determine the conversion of benzaldehyde and the selectivity to the target product of benzaldehyde ethylene glycol acetal. In order to investigate the beginning of active sites, the catalytic reaction on Fe3O4@C-600 without sulfonation treatment was studied at first (Table 1), which showed almost no catalytic activity toward benzaldehyde conversion, further confirming that the acetalization reaction is conducted at the acid sites of sulfonated catalyst. According to the description above, through controlled carbonization at different temperatures from 300 ℃ to 600 ℃, the Fe3O4@C microspheres with tunable graphitization degrees of carbon shells were chemically grafted with SO3H groups. The obtained Fe3O4@C-SO3H catalysts showed gradually increased acid amount in the range of 0.59–1.04 mmol/g, resulting in improved conversion of benzaldehyde from 49% to 69% for the catalysts of Fe3O4@C-x-SO3H in sequence (x=300, 400, 500, 600) (Table 1 and Fig. 5b). In addition, the catalyst of Fe3O4@C-700-SO3H presented the lowest benzaldehyde conversion (31%), corresponding to the lowest acid amount among all the sulfonic acid-functionalized Fe3O4@C microspheres. Since Fe3O4@C-600-SO3H possessed superparamagnetic property, the highest acid amount (1.04 mmol/g) and catalytic activity in acetalization reaction, its reaction process and recyclability were studied in detail. The conversion of benzaldehyde increases with the reaction time, reaching the maximum value of 69% at 2h (Fig. 6a), which is higher than the reported values on other acid catalysts [48-51]. For comparison, the C-600-SO3H without a magnetic core was also studied (Table 1), in which lower acid amount (0.38 mmol/g) resulted in lower conversion of benzaldehyde (27%) than any of the Fe3O4@C-x-SO3H catalysts. This is mainly attributed to the structural expansion of RF and carbon shells by the inner Fe3O4 cores in Fe3O4@C microspheres, thus providing abundant active sites for further sulfonation. During the reaction, Fe3O4@C-600-SO3H always keeps high selectivity to benzaldehyde ethylene glycol acetal, which gets to the highest point of 97% at 2 h, suggesting that Fe3O4@C-600-SO3H performs as a superior catalyst for the acetalization reaction. For the magnetic solid acid catalyst, feasible recyclability is very important for reusing the catalyst in practical catalytic reaction, which was evaluated on Fe3O4@C-600-SO3H under the same conditions. As shown in Fig. 6b, the conversion of benzaldehyde is quite stable within 9 cycles with a slow decrease from 69% to 64.8%, revealing the outstanding recyclability of Fe3O4@C-600-SO3H only through a convenient and fast separation under a magnetic field for successive cycles. All the results above prove that Fe3O4@C-600-SO3H is a promising magnetically recyclable solid acid catalyst, and the favorable catalytic properties stem from its novel magnetic core-shell structure containing rigid carbon shells of high graphitization degree, superparamagnetic property and a great number of stably-grafted sulfonic acid groups. The magnetic solid acid catalyst may also be feasible in catalyzing other similar acetalization reactions including common reactants such as formaldehyde, acetone, etc., which further broadens its practical applications in acid-catalytic reactions.

|
Download:
|
| Fig. 5. (a) Acid-catalyzed acetalization reaction between benzaldehyde and ethylene glycol. (b) Acid amounts and conversions of benzaldehyde on different Fe3O4@C-x-SO3H catalysts (x=300, 400, 500, 600 and 700). | |
{kind=link}

|
Download:
|
| Fig. 6. (a) Conversion of benzaldehyde (red line) and selectivity to benzaldehyde ethylene glycol acetal (black line) vs. reaction time on Fe3O4@C-600-SO3H as the catalyst. (b) Recyclability of Fe3O4@C-600-SO3H in acetalization reaction between benzaldehyde and ethylene glycol. | |
{kind=link}
In conclusion, a versatile interfacial polymerization combined with a controlled sulfonation treatment strategy was employed for rational design and synthesis of magnetic core-shell Fe3O4@C-x-SO3H solid acid catalysts. The controllable interfacial polymerization between resorcinol and formaldehyde using weak alkaline ammonia solution facilitated the coating of RF layers in tunable thickness, and the regulation of graphitization degree of carbon shells showed a close relationship with the surface acid amounts of sulfonated microspheres. The core-shell structure showed quite well thermodynamic stability during carbonization process, and moderate carbonization temperature realized high graphitization degree and increased surface acid amounts as well, further promoting catalytic activities. Among the magnetic core-shell Fe3O4@C-x-SO3H solid acid catalysts, Fe3O4@C-600-SO3H possessed relatively high conversion (69%) and selectivity (97%) for the acetalization reaction between benzaldehyde and ethylene glycol. Moreover, the catalysts showed excellent magnetic recyclability due to their unique superparamagnetic properties and stability of grafted sulfuric acid sites. It is expected that such core-shell magnetic solid acid catalysts can be widely applied in organic synthesis to realize a green and sustainable chemical process.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21875044, 52073064, 22005058 and 22005057), National Key R & D Program of China (No. 2020YFB2008600), Key Basic Research Program of Science and Technology Commission of Shanghai Municipality (No. 20JC1415300), Program of Shanghai Academic Research Leader (No. 19XD1420300), China Post-doctoral Science Foundation (Nos. 2020M670973, BX20200085), and the State Key Laboratory of Transducer Technology of China (No. SKT1904). The authors extend their thanks to Research Supporting Project (No. RSP-2020/155), King Saud University, Riyadh, Saudi Arabia.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.027.
| [1] |
N. Hidayati, T. Pujiati, E.B. Prihandini, et al., Bull. Chem. React. Eng. Catal. 14 (2019) 683-688. DOI:10.9767/bcrec.14.3.4254.683-688 |
| [2] |
D.J. Kim, M.J. Jo, S.Y. Nam, J. Ind. Eng. Chem. 21 (2015) 36-52. DOI:10.1016/j.jiec.2014.04.030 |
| [3] |
A.R. Kim, M. Vinothkannan, D.J. Yoo, Int. J. Hydrogen Energy 42 (2017) 4349-4365. DOI:10.1016/j.ijhydene.2016.11.161 |
| [4] |
S.S. Araya, F. Zhou, V. Liso, et al., Int. J. Hydrogen Energy 41 (2016) 21310-21344. DOI:10.1016/j.ijhydene.2016.09.024 |
| [5] |
A.C. Dupuis, Prog. Mater. Sci. 56 (2011) 289-327. DOI:10.1016/j.pmatsci.2010.11.001 |
| [6] |
M. Amirinejad, S.S. Madaeni, E. Rafiee, et al., J. Membr. Sci. 377 (2011) 89-98. DOI:10.1016/j.memsci.2011.04.014 |
| [7] |
A. Chandan, M. Hattenberger, A. El-kharouf, et al., J. Power Sources 231 (2013) 264-278. DOI:10.1016/j.jpowsour.2012.11.126 |
| [8] |
A. Kraytsberg, Y. Ein-Eli, Energy Fuels 28 (2014) 7303-7330. DOI:10.1021/ef501977k |
| [9] |
Y. Zhai, Y. Dou, D. Zhao, et al., Adv. Mater. 23 (2011) 4828-4850. DOI:10.1002/adma.201100984 |
| [10] |
M. Cossutta, V. Vretenar, T.A. Centeno, et al., J. Clean. Prod. 242 (2020) 118468. DOI:10.1016/j.jclepro.2019.118468 |
| [11] |
J. Wang, Y. Xia, Y. Liu, et al., Energy Stor. Mater. 22 (2019) 147-153. DOI:10.1016/j.ensm.2019.01.008 |
| [12] |
X. Huang, K. Qian, J. Yang, et al., Adv. Mater. 24 (2012) 4419-4423. DOI:10.1002/adma.201201680 |
| [13] |
Y. Fang, Y. Lv, R. Che, et al., J. Am. Chem. Soc. 135 (2013) 1524-1530. DOI:10.1021/ja310849c |
| [14] |
J. Meng, C. Niu, L. Xu, et al., J. Am. Chem. Soc. 139 (2017) 8212-8221. DOI:10.1021/jacs.7b01942 |
| [15] |
M. Hara, T. Yoshida, A. Takagaki, et al., Angew. Chem. Int. Ed. 43 (2004) 2955-2958. DOI:10.1002/anie.200453947 |
| [16] |
M. Toda, A. Takagaki, M. Okamura, et al., Nature 438 (2005) 178. DOI:10.1038/438178a |
| [17] |
M. Okamura, A. Takagaki, M. Toda, et al., Chem. Mater. 18 (2006) 3039-3045. DOI:10.1021/cm0605623 |
| [18] |
K. Nakajima, M. Okamura, J.N. Kondo, et al., Chem. Mater. 21 (2009) 186-193. DOI:10.1021/cm801441c |
| [19] |
S. Suganuma, K. Nakajima, M. Kitano, et al., J. Am. Chem. Soc. 130 (2008) 12787-12793. DOI:10.1021/ja803983h |
| [20] |
K. Nakajima, M. Hara, ACS Catal. 2 (2012) 1296-1304. DOI:10.1021/cs300103k |
| [21] |
M. Hara, Energy Environ. Sci. 3 (2010) 601-607. DOI:10.1039/b922917e |
| [22] |
X. Zhang, Y. Zhao, S. Xu, et al., Nat. Commun. 5 (2014) 3170-3178. DOI:10.1038/ncomms4170 |
| [23] |
X. Zhang, L. Zhang, Q. Yang, J. Mater. Chem. A 2 (2014) 7546-7554. DOI:10.1039/C4TA00241E |
| [24] |
X. Zhang, Y. Zhao, Q. Yang, J. Catal. 320 (2014) 180-188. DOI:10.1016/j.jcat.2014.09.018 |
| [25] |
V. Polshettiwar, R. Luque, A. Fihri, et al., Chem. Rev. 111 (2011) 3036-3075. DOI:10.1021/cr100230z |
| [26] |
A. Lu, E.L. Salabas, F. Schüth, Angew. Chem. Int. Ed. 46 (2007) 1222-1244. DOI:10.1002/anie.200602866 |
| [27] |
P. Tartaj, M.P. Morales, T. González-Carreño, et al., J. Magn. Magn. Mater. 290 (2005) 28-34. |
| [28] |
J. Gao, H. Gu, B. Xu, Acc. Chem. Res. 42 (2009) 1097-1107. DOI:10.1021/ar9000026 |
| [29] |
S. Shylesh, V. Schünemann, W.R. Thiel, Angew. Chem. Int. Ed. 49 (2010) 3428-3459. DOI:10.1002/anie.200905684 |
| [30] |
Z. Tai, M.A. Isaacs, C.M.A. Parlett, et al., Catal. Commun. 92 (2017) 56-60. DOI:10.1016/j.catcom.2017.01.004 |
| [31] |
M. Zhu, G. Diao, Nanoscale 3 (2011) 2748-2767. DOI:10.1039/c1nr10165j |
| [32] |
W. Liu, K. Tian, H. Jiang, et al., Sci. Rep. 3 (2013) 2419-2425. DOI:10.1038/srep02419 |
| [33] |
H. Guo, Y. Lian, L. Yan, Smith, et al., Green Chem. 15 (2013) 2167-2174. DOI:10.1039/c3gc40433a |
| [34] |
Q. Yue, J. Li, W. Luo, et al., J. Am. Chem. Soc. 137 (2015) 13282-13289. DOI:10.1021/jacs.5b05619 |
| [35] |
Q. Yue, J. Li, Y. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 15486-15493. DOI:10.1021/jacs.7b09055 |
| [36] |
Q. Yue, Y. Zhang, Y. Jiang, et al., J. Am. Chem. Soc. 139 (2017) 4954-4961. DOI:10.1021/jacs.7b01464 |
| [37] |
L. Wan, H. Song, X. Chen, et al., Adv. Mater. 30 (2018) 1707515. DOI:10.1002/adma.201707515 |
| [38] |
Y. Zhang, Q. Yue, M.M. Zagho, et al., ACS Appl. Mater. Interfaces 11 (2019) 10356-10363. DOI:10.1021/acsami.8b18721 |
| [39] |
L. Yu, P. Pan, Y. Zhang, et al., Small 15 (2019) 1805465. DOI:10.1002/smll.201805465 |
| [40] |
P. Pan, T. Zhang, Q. Yue, et al., Adv. Sci. 7 (2020) 2000443. DOI:10.1002/advs.202000443 |
| [41] |
Y. Zhu, T. Ikoma, N. Hanagata, et al., Small 6 (2010) 471-478. DOI:10.1002/smll.200901403 |
| [42] |
M. Wang, Y. Ni, A. Liu, ACS Omega 2 (2017) 1505-1512. DOI:10.1021/acsomega.7b00064 |
| [43] |
P. Tan, Y. Jiang, X. Liu, et al., ACS Sustain. Chem. Eng. 4 (2016) 2223-2231. DOI:10.1021/acssuschemeng.5b01713 |
| [44] |
J. Liu, Z. Sun, Y. Deng, et al., Angew. Chem. Int. Ed. 48 (2009) 5875-5879. DOI:10.1002/anie.200901566 |
| [45] |
S.A. Al-Muhtaseb, J.A. Ritter, Adv. Mater. 15 (2003) 101-114. DOI:10.1002/adma.200390020 |
| [46] |
L. Wang, K. Gan, D. Lu, et al., Eur. J. Inorg. Chem. (2016) 890-896. |
| [47] |
Z. Chen, Z. Geng, Z. Zhang, et al., Eur. J. Inorg. Chem. (2014) 3172-3177. |
| [48] |
P. Srivastava, R. Srivastava, Catal. Commun. 9 (2008) 645-649. DOI:10.1016/j.catcom.2007.07.008 |
| [49] |
Z. Duan, Y. Gu, Y. Deng, Catal. Commun. 7 (2006) 651-656. DOI:10.1016/j.catcom.2006.02.008 |
| [50] |
B. Wang, Y. Gu, G. Song, et al., J. Mol. Catal. A Chem. 233 (2005) 121-126. DOI:10.1016/j.molcata.2005.02.006 |
| [51] |
H. Wu, F. Yang, P. Cui, et al., Tetrahedron Lett. 45 (2004) 4963-4965. DOI:10.1016/j.tetlet.2004.04.138 |