 2021, Vol. 32
2021, Vol. 32 


b Sino-German Centre for Water and Health Research, Sichuan University, Chengdu 610065, China;
c Department of Central Transportation Center, West China Hospital, Sichuan University/West China School of Nursing, Sichuan University, Chengdu 610065, China;
d Institute of Environmental Engineering, RWTH Aachen University, Aachen 52072, Germany;
e Sichuan University Yinbin Park, Yibin Institute of Industrial Technology, Yibin 644044, China;
f Laboratory of Wastewater Treatment Technology in Sichuan Province, Haitian Water Group, Chengdu 610065, China
As typical nitroaromatic compounds, p-nitrophenol (PNP) and its derivatives are extensively used in plasticizer, pesticide, medicine, dyes, and other high-pollution chemical industry. Previous studies had demonstrated that PNP can stimulate and inhibit the central nervous system, even causing dyspnea and hyperhemoglobinemia [1]. Besides, due to its high toxicity, water-solubility, excellent chemical and biological stability, PNP had been listed as a priority pollutant by the US Environmental Protection Agency (EPA) [2].
To remove PNP efficiently from water bodies, a series of approaches have been studied, such as physical adsorption [3], biodegradation [4], and chemical degradation [5, 6]. Especially, advanced oxidation processes (AOPs) have been widely applied by catalyzing the generation of highly active free radicals which could oxidize PNP into low toxicity of small molecules. In decades, peroxymonosulfate (PMS), persulfate (PDS), hydrogen peroxide (H2O2) have been increasingly considered in the AOPs own to their superior oxidation performance [7, 8]. They could be activated by UV light, transition metal/metal oxidizers, and ultrasound to generate sulfate radical (SO4·−) and hydroxyl radical (HO·) [9-12].
Several studies had demonstrated that zero-valent iron (ZVI) was considered as high-efficient and applicable activators for PDS/PMS/H2O2 seeing that its low cost, environmentally friendly, and high reductive capacity (Eqs. 1-4) [13-17]. Yang et al. found that methyl orange could be degraded efficiently in the ZVI/PDS system [18], and Wang et al. reported that both ZVI/PMS and ZVI/H2O2 systems could decompose azo dye maximumly [19]. In our previous study, efficient removal of high concentration PNP could maintain by the ZVI with ultrasonic irradiation (US-ZVI) system [20], and we also found that microscale Fe/Cu bimetallic particles with PDS could remove PNP approached 100% in aqueous solution [21]. However, the excess oxidizer improved producing Fe2+ of ZVI corrosion products, which would scavenge the generated radicals (Eqs. 5 and 6) [22, 23]. An interesting phenomenon also could be obtained that the oxidizers, generated free radicals and produced H+ with the dissolution of the oxidizers could accelerate ZVI corrosion [24, 25]. Surprisingly, there is no report on the contribution of oxidizers to enhancing the reducibility of ZVI during the co-treatment process of contaminants.
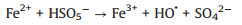
|
(1) |
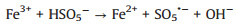
|
(2) |
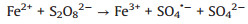
|
(3) |
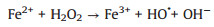
|
(4) |

|
(5) |
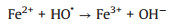
|
(6) |
Hence, the objective of this study was to confirm the synergistic mechanisms between ZVI and oxidizers. The effects of oxidizer dosages (PDS, PMS and H2O2) on undegraded ratio and oxidative degradation ratios, especially the reductive degradation ratio of PNP during reaction systems were comparatively analyzed. The contribution of promoting/inhibition effect specifically of produced ROS or H+ under different oxidizer dosage on reductive ratios reaction was ascertained. To pinpoint the degradation properties of PNP in different systems that were under the strongest reductive conditions by analyzing the concentration curves of intermediates, TOC removal and UV–vis absorption spectra.
In previous studies, the mechanisms for PNP removal by ZVI were described thoroughly [26, 27]. As shown in Fig. 1, two degradation pathways of PNP based on the above work were proposed. On the one hand, PNP could be reduced to PAP (p-aminophenol) by ZVI or Fe2+ directly, and then which would be oxidized to HQ (hydroquinone) and BQ (p-benzoquinone) by ROS. On the other hand, PNP could be oxidized directly to HQ and BQ. In consequence, PAP as a characteristic reductive intermediate during the reduction process of the nitro group of PNP was selected to represent the ratio of reduction in the treatment process. The real-time reductive ratio was equal to the molar concentration of generated PAP divided by the initial molar concentration of PNP. And the rest proportion was the oxidative degradation ratio.

|
Download:
|
| Fig. 1. Main reaction pathways for the degradation of PNP. | |
{kind=link}
Figs. 2a and b show the variation of the undegraded PNP, oxidative degradation, and reductive degradation ratios with the increase of the molar ratio of ZVI to PDS ([ZVI]: [PDS]) in the ZVI/PDS system. With the increase of the molar ratio of ZVI to PDS from 0 to 100:1, the PNP removal efficiency increased from 26.5%–98.7% rapidly after 15 min treatment. It could be observed that the reductive ratio was significantly increased from 25.6% to maximum (82.2%) when the molar ratio of ZVI to PDS reached 100:1. The oxidative degradation ratio of PNP continued to increase with the increase of PDS dosages, and 16.5% could be obtained when [ZVI]: [PDS] was 100:1.

|
Download:
|
| Fig. 2. The ratios of undegraded PNP, oxidative degradation, reductive degradation after 5 min in ZVI/PDS (a), ZVI/PMS (c), ZVI/H2O2 (e) system and 15 min in ZVI/PDS (b), ZVI/PMS (d), ZVI/H2O2 (f) system (Inset: the changing trends of oxidative and reductive ratios with the increase of oxidizer dosages). Experimental conditions: [PNP]0=100 mg/L, [ZVI]0=10 g/L, T=25±1 ℃, initial pH 5.76. | |
{kind=link}
Similar results were also obtained in the ZVI/PMS and ZVI/H2O2 systems. As shown in Figs. 2c and d, PNP removal efficiency increased to 99.3% rapidly when the molar ratio of ZVI to PMS reached 100:1, while the reductive ratio reached the peak value of 83.4%. Figs. 2e and f depicts the variation of the three parts by increasing the H2O2 dosage in the ZVI/H2O2 system. Only 62.2% PNP was removed when the molar ratio of ZVI to H2O2 increased to 1:1. The highest reductive degradation efficiency was 46.1% when the molar ratio of ZVI to H2O2 was 100:1.
The unified phenomena in these three systems reflected that the main degradation process of PNP is the gradual replacement of the reduction process by the oxidation process with the increase of oxidizer dosages. The variation trends of PNP removal could be attributed to the generated ROS and the scavenging effect of excess Fe2+ on ROS [22, 23]. With the increase of generated ROS and the consumption of H+ during the reaction, the oxidative ratio increased significantly after 15 min compared to 5 min. Moreover, it could be observed that the tendency to increase first and then decrease of reductive degradation ratio with the increase of oxidizer dosage. The nonnegligible enhancement of reductive degradation in these co-systems compared to the ZVI alone system, which might be attributed to the released H+ and generated ROS after the addition of oxidizers. On this basis, the effects of pH variation and generated ROS on reductive degradation of PNP were investigated thoroughly, respectively.
Generally, organic contaminants could be degraded efficiently by PDS/PMS systems over a wide range of pH [28]. In comparison, the Fenton-like system (ZVI/H2O2) was intensely dependent on initial pH [29]. Furthermore, the addition of PDS/PMS would cause a significant variation of the initial pH in solution while a relatively constant pH occurred after adding H2O2. Figs. S1a, c and Figs. S1b, d (Supporting information) show the PNP reductive degradation ratios after 5 min, 15 min under different pH, and under different molar addition ratios of ZVI/PDS and ZVI/PMS systems, respectively. Control experiments were carried by only initial pH adjustment (pH0 1.5–5.5) in the ZVI system to ascertain the contribution of H+ to reduction. As shown in Fig. S1c, almost half of the maximum reductive ratio in the ZVI/PDS system was attributed to the promotion of H+. In the ZVI/PMS system (shown in Fig. S1b), near 58.0% of the reductive ratio could be achieved under the influence of H+ released by PMS while the maximum reductive ratio (([ZVI]: [PMS]=100:1) was 83.3%. The results verified the great role of H+ in promoting the reductive reaction.
The evolution of solution pH in ZVI/PDS and ZVI/PMS systems under different PDS or PMS dosage were illustrated in Figs. S2a and b (Supporting information). The solution pH of the ZVI/PDS system decreased sharply within the initial 1 min, and the solution pH of the ZVI/PMS system decreased to the minimum immediately after adding PMS. The phenomena were on account of the formation of plenty HSO4−, which formed by the hydrolysis of PDS/PMS and the reaction of SO4·− with H2O in solution simultaneously (Eqs. 7-10) [30, 31]. During the reaction process, the solution pH increased distinctly and approached to a flat. In comparison to ZVI/PDS system, the faster reactive balance could be observed in the ZVI/PMS system. Of note is the slight decrease occurred at solution pH in both two systems ultimately while less rangeability in the ZVI/PMS system. This variation tread of solution pH of the ZVI/PDS system was consistent with that of the ZVI/PMS system. One of the reasons for the phenomenon is the consumption of H+ during the corrosion of ZVI. Another is that the generated small molecule intermediates (e.g., fumaric acid and maleic acid) from the degradation of PNP would contribute the H+ [32]. Furthermore, there was no change in the ZVI/PMS system (([ZVI]: [PMS]=100:1) ultimately compared with the descend range of 0.8% in the ZVI/PDS system (([ZVI]: [PDS]=100:1). The result might imply more production of small molecule acids in the ZVI/PDS system.
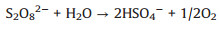
|
(7) |
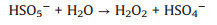
|
(8) |
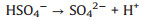
|
(9) |
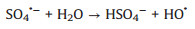
|
(10) |
To comprehensively evaluate the effects of ROS produced under different reaction systems on the reductive ratio, the quenching experiments under the condition of the maximum reductive ratio in each reaction system were carried out. The results were shown in Figs. S3–S5 (Supporting information). According to the differences in rate constants between radicals and scavengers, EtOH and TBA were selected as the radical scavengers with the addition of 100 mmol/L, respectively. EtOH easily reacts with both HO· (1.2×109 – 2.8×109 L mol−1 s−1) and SO4·− (1.6×107 – 7.7×107 L mol−1 s−1), while TBA has much higher rate constant with HO· (3.8×108 – 7.6×108 L mol−1 s−1) than SO4·− (4.0×105 – 9.1×105 L mol−1 s−1) [33, 34]. As shown in Figs. S3 and S4, for ZVI/PDS and ZVI/PMS systems, when the molar ratio of ZVI: PDS was 500:1 and ZVI: PMS was 1000:1, the reductive ratio dropped from 46.3%, 41.9%–39.7%, 29.5% by quenching experiments, respectively. However, when the molar ratio further increased to 200:1 in ZVI/PDS system and 100:1 in ZVI/PMS system, the promoting effect of ROS with on the reductive reaction turned into inhibition ultimately. The above results indicate that the generated ROS could effectively enhance the reduction capacity of ZVI particles when the low molar ratio of ZVI to oxidizer was adopted during the reaction. Of note is the significant increase occurred in the accelerative effect of ROS on reductive reaction after 5 min in each system. This might imply that the promotion to the reduction in the first 5 min was mainly caused by generated H+, which was also consistent with the rapid increase of pH during the reaction over the first 5 min.
In general, at the same molar concentration of oxidants, the reducibility of ZVI follows the trend that PMS system > PDS system > H2O2 system. Under the same conditions, PMS would hydrolyze more H+ to promote the corrosion of ZVI strengthening the reductive degradation compared to PDS [31]. Meanwhile, the activation of oxidizers to produce ROS would also be enhanced [13-17].
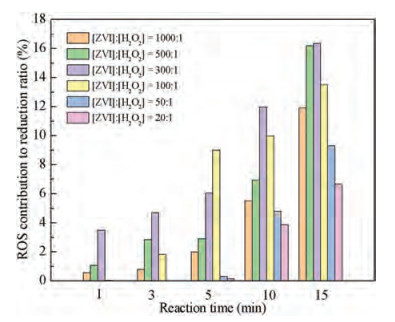
Different from the persulfate systems, there was no interference of H+ in the ZVI/H2O2 system. As shown in Fig. S5, the promotion to the reductive reaction of the produced ROS could be observed consistently in ZVI/H2O2 system even under the condition of high H2O2 dosage ([ZVI]: [H2O2]=20:1). This could be ascribed to the production of fewer free radicals due to few H+ (Eqs. 11 and 12) [35, 36]. Based on the quenching experiments of the ZVI/H2O2 system, the contribution of ROS to promoting reductive reaction during 15 min process under different molar ratios in the ZVI/H2O2 reaction system at initial pH of 5.76 (the pH of original solution) was exhibited in Fig. 3. The specific contribution was obtained by the calculation that the reductive degradation ratio of quenching was divided by the original. And the reductive degradation ratio of quenching was equal to the original reduction degradation ratio minus the reduction degradation ratio after quenching (Eq. 13). On one hand, it could be observed that the contribution of ROS to the reductive reaction increased with the reaction time. And on the other hand, the contribution of ROS to reduction increased at the outset and then decreased with the increase of H2O2 dosage. More specifically, the contribution of ROS reached a maximum of 16.3% when the molar ratio of ZVI to H2O2 reached to 300:1 after 15 min treatment. Additionally, there was still a promoting contribution of 6.6% even the molar ratio of ZVI to H2O2 increased to 20:1 after 15 min treatment.
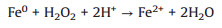
|
(11) |

|
(12) |

|
(13) |

|
Download:
|
| Fig. 3. The contribution of ROS to promoting reductive reaction during 15 min process under different molar ratios in the ZVI/H2O2 system. Experimental conditions: [PNP]0=100 mg/L, [ZVI]0=10 g/L, T=25±1 ℃, initial pH 5.76. | |
{kind=link}
The total iron ions leaching in different treatment systems was shown in Fig. S6 (Supporting information), and the detailed discussion was stated in Text S1 (Supporting information).
Figs. S7a–c (Supporting information) shows the TOC removal efficiencies obtained by different reaction systems. Only 3.27% of the TOC removal was obtained after 15 min treatment in ZVI alone system, and the TOC removal efficiency increased obviously with the addition of oxidizer. In the ZVI/PDS system, 6.24%, 7.72%, 17.56%, 33.78% and 55.42% of TOC removal could be achieved with the molar ratio of ZVI to PDS increased gradually from 500:1 to 200:1, 50:1, 20:1 and 3:1, respectively. A similar variation was observed in the ZVI/PMS system. Smaller increments occurred in the ZVI/H2O2 system, the maximum of TOC removal efficiency was 12.78% when the molar ratio of ZVI to H2O2 was 1:1. The above phenomena could be ascribed to the increasing generated ROS with the increase of oxidizer dosages, which was consistent with the increased oxidative degradation of PNP in Fig. 1. Nonetheless, when the reductive degradation ratio of PNP in each reaction system reached the maximum ([ZVI]: [PDS] = [ZVI]: [PMS] = [ZVI]: [H2O2]=100:1), the TOC removal efficiencies were only 10.75%, 13.49% and 8.32% of ZVI/PDS, ZVI/PMS and ZVI/H2O2, respectively. The incomplete mineralization rates of PNP indicated that the existence of intermediates possibly after the treatment.
Figs. 4a-c show molar concentration profiles of PNP and the primary intermediates (e.g., PAP, HQ, BQ) which were detected by HPLC analysis. As shown in Fig. 4a, it was observed that the BQ concentration rapidly increased to the maximum concentration within the first 1 min in the ZVI/PDS system, but nearly decomposed after 3 min later. The concentration of PAP and HQ did not increase significantly in the process of the rapid increase of the BQ, and both of them increased dramatically while the BQ concentration decreased. Similar trends were found in the ZVI/PMS system. Besides, the concentration of HQ increased rapidly and remained at about 0.05 mmol/L, 0.11 mmol/L after 3 min in ZVI/PDS and ZVI/PMS systems, respectively. It also could be observed that the decrease of BQ concentration synchronized with the increase of HQ concentration. Furthermore, good molar balance could be seen in these three systems, implying that PAP, HQ and BQ were the primary intermediates in these systems. However the balances never achieved 100%, which indicates that some products of PNP might be absorbed onto the ZVI suaface or their corrosion products, and/or additional intermediates such as formic and maleic acids [37]. In addition, reproducible dips occurred at the initial stage of reaction in three systems. This might be attributed to absorbing firstly of PNP molecules by ZVI surface and then degrading. And/or PNP was reduced to p-nitrosophenol before being reduced to PAP, which was not detected during the analysis process [38]. A similar phenomenon was also found in our previous study [39]. The above results demonstrate that PNP was mainly degraded by oxidation as the dominant position with a slight reductive reaction in the first 1 min. It might be attributed to the rapid formation of free radicals. And after 1 min, the reductive reaction quickly replaced the oxidation reaction as the dominant reaction with the corrosion of ZVI.Fig. 4c revealed that the dominant oxidation reaction at the initial 1 min and the subsequent main reduction in degradation also occurred in the ZVI/H2O2 system (pH0 5.76). However, lower level in oxidation and reduction could be observed compared with ZVI/PDS and ZVI/PMS systems, which might be ascribed to fewer free radicals in the ZVI/H2O2 system.

|
Download:
|
| Fig. 4. Molar concentration profiles of PNP and the intermediates over time in different systems: ZVI/PDS (a); ZVI/PMS (b); ZVI/H2O2 (c). Experimental conditions: [ZVI]: [PDS] = [ZVI]: [PMS] = [ZVI: [H2O2]=100:1, [PNP]0=100 mg/L, [ZVI]0=10 g/L, T=25±1 ℃, initial pH 5.76. | |
{kind=link}
The UV–vis spectra from 190 nm to 500 nm of the influent and effluent of ZVI/PDS, ZVI/PMS, ZVI/H2O2 systems were shown in Fig. 5. Based on our previous study, the peaks at 227 nm and 317 nm are mainly attributed to the π–π* transition of the benzene ring of monoaromatics and the conjugation of the benzene ring and chromophore group (e.g., -NO2), respectively [40]. A similar phenomenon could be observed in three different systems that absorbance intensity of the peak at 317 nm decreased rapidly differ from that at 227 nm. The phenomena reveal that the-NO2 on the molecular structure of PNP could be reduced easily, however, only part of the benzene ring of PNP could be oxidized to open the ring and further mineralized. Furthermore, a slight decrease occurred at both absorbance intensity of the peak at 317 nm and 227 nm in the ZVI alone system, which also illustrated that the addition of an oxidizer could promote the oxidation and reduction reaction. A new peak at 273 nm emerged in these three systems, also suggesting that the -NO2 of PNP was reduced into -NH2 result in the formation of PAP. Meanwhile, the transition from the absorption intensity peak at 273 nm to the absorption intensity peak at 246 nm also suggested the formation of BQ. In addition, the decrease in absorbance intensity of the peak at 317 nm was most dramatic in the ZVI/PMS system, followed by the ZVI/PDS system and ZVI/H2O2 system, respectively. The above results are following the trend of TOC removal efficiency in these three treatment systems.

|
Download:
|
| Fig. 5. UV–vis spectra of the effluent after 15 min and time-course variations of UV–vis spectra during 15 min process under the condition of the strongest reductive degradation of ZVI/PDS system (a, b); ZVI/PMS system (c, d); ZVI/H2O2 system (e, f). Experimental conditions: [PNP]0=100 mg/L, [ZVI]0=10 g/L, T=25±1 ℃, initial pH 5.76. | |
{kind=link}
As shown in Figs. S8a–f (Supporting information), the surface morphology and elemental composition of the reacted ZVI particles in different systems were characterized by SEM and EDS. Compared to ZVI alone system, more obvious oxidation corrosion products could be seen on the surface of reacted ZVI particles. More specifically, Table S1 (Supporting information) shows that nearly 10.5 wt% of O and 89.5 wt% of Fe were detected on the surface of reacted ZVI particles in the ZVI alone system. Additionally, nearly 31.2 wt%, 30.8 wt%, and 24.4 wt% of O on the surface of reacted ZVI particles in ZVI/PDS, ZVI/PMS, and ZVI/H2O2 systems could be obtained, respectively, while almost 68.8 wt%, 69.1 wt% and 75.6 wt% of Fe were detected in these three systems. The above results suggest that the addition of oxidizers could enhance the corrosion of ZVI to promote the oxidation and reduction of PNP effectively. Meanwhile, the lower mass fraction ratio of O and higher of Fe in the ZVI/H2O2 system also verified the stronger corrosion of ZVI in PDS/PMS system by the co-promotion of H+ and ROS.
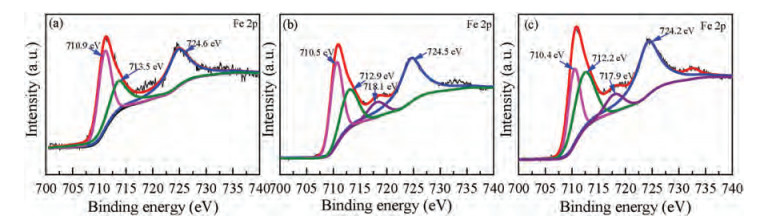
To identify the chemical state of Fe and O of reacted ZVI particles under the condition of the strongest reductive degradation, XPS was carried after the treatment of different systems. All XPS core-level spectra were fitted using Shirley background. As shown in Figs. 6a-c, further detailed scans in different systems have performed for Fe 2p core-level spectra to determine the charge state of elements present on the surface of reacted ZVI. It had been reported that the binding energies at 710.0–720.0 eV for iron oxides could be attributed to Fe 2p3/2 [41].The peaks at 710.9 eV, 710.5 eV and 710.4 eV in these three treatment systems were all close to Fe3+ state in Fe2O3 at 710.7 eV [42, 43]. Furthermore, the signals of Fe 2p3/2 binding energy position were observed at 712.9 eV and 712.2 eV in FeOOH, while Fe 2p1/2 binding energy position was observed at 724.6 eV, 724.5 eV and 724.2 eV in Fe2O3 [44, 45]. Of note is the observed peak at 713.5 eV which corresponded to the reaction product of SO42− in FeSO4 [46].

|
Download:
|
| Fig. 6. Fe 2p core level of XPS spectra of the reacted ZVI in ZVI/PDS system (a); ZVI/PMS system (b); ZVI/H2O2 system (c). Experimental conditions: [ZVI]: [PDS] = [ZVI]: [PMS] = [ZVI]: [H2O2]=100:1, [PNP]0=100 mg/L, [ZVI]0=10 g/L, T=25±1 ℃, initial pH 5.76. | |
{kind=link}
The high binding energies of 718.1 eV and 717.9 eV observed between the Fe 2p3/2 and Fe 2p1/2 also could be assigned to Fe2+. The full-range scans and O 1 s core level of XPS spectroscopy of the reacted ZVI in different systems were shown in Figs. S9a–c and Figs. S10a-S10c (Supporting information), respectively. And the detailed analysis of the O 1 s core level is provided in the Text S2. The results further reveal that a few iron corrosion products (Fe2O3, Fe3O4, Fe(OH)2, Fe(OH)3 and FeOOH) could be deposited on the surface of reacted ZVI particles, which was consistent with SEM analysis. Moreover, these (hydr)oxides could further activate PDS/PMS/H2O2 to enhance the generation of ROS and the degradation of PNP.
The major reactions and removal mechanisms of PNP degradation in ZVI/PDS, ZVI/PMS, and ZVI/H2O2 systems were proposed. Firstly, the released H+ from the hydrolysis of oxidizers and raw wastewater could accept the released electrons from iron corrosion and form free hydrogen under acid condition ([H]), which would rapidly reduce PNP [47]. H+ could also accelerate the corrosion rate of ZVI. For another, oxidizers could directly oxidize ZVI in situ to promoting the release Fe2+ which also could be accelerated by ZVI corrosion under the promotion of generated ROS [48]. Thus, PNP could directly accept the released electrons and be reduced to PAPin situ, Fe2+ in solution also might cause the reductive reaction of PNP [49, 50]. As a result, the addition of oxidizers could enhance reductive degradation during the degradation of PNP. The addition of oxidizers could also enhance the oxidic effect and improve the removal efficiency of PNP. On one hand, the generated Fe2+/ Fe3+ could facilitate the activation of oxidizers to produce ROS (SO4·−, HO· and SO5·−) which would oxidize PNP or PAP to form HQ, BQ, even CO2, and H2O ultimately. On another hand, Fe2+/Fe3+ could readily transform to iron hydroxides (Fe(OH)2, Fe(OH)3) as the raising pH during the reaction, which could remove PNP and its intermediates through adsorption and co-precipitation [51, 52]. Furthermore, the generated corrosion products (Fe2O3, Fe3O4 and FeOOH) could further activate oxidizers to generate ROS in return [53, 54].
This paper discussed the mechanism of oxidizers enhancing PNP degradation in the presence of ZVI. Monitoring the effects of different single oxidizer dosages on the ratios of oxidative and reductive degradation of PNP, it was found that the main degradation process of PNP was the gradual replacement of the reduction process by the oxidation process with the increase of oxidizer dosages. Control experiments with only pH adjustment and no oxidizer added confirmed the remarkable promotion of H+ released by oxidizers on reductive reaction. Quenching experiments also revealed that ROS produced by small oxidizer dosage could accelerate the reduction of PNP. Take the case of the ZVI/H2O2 system, the maximum of 16.3% could be obtained which was the contribution to ROS to reductive degradation of PNP. Based on the presence of PAP in large quantities during the reaction process and the results of UV–vis absorption spectra and intermediates concentration curves, PNP was mainly degraded by reduction accompanied by slight oxidation when the molar ratio of ZVI to oxidizer less than or equal to 10. Besides, the generated ROS, the additional H+, and the corrosion products of ZVI promoted PNP degradation based on the analysis of SEM-EDS and XPS. This work investigated the specific promoting mechanisms and contribution after the addition of oxidizers on the corrosion of ZVI thoroughly.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThe authors would like to acknowledge the financial support from Key R & D projects of the Sichuan Science and Technology Plan Project (No. 19ZDYF0612), Guiding Plan for Transfer and Transformation of Scientific and Technological Achievements of Sichuan Science and technology plan project (No. 20ZHSF0257), and the National Natural Science Foundation of China (No. 51878423).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.019.
| [1] |
G. Eichenbaum, M. Johnson, D. Kirkland, et al., Regul. Toxicol. Pharmacol. 55 (2009) 33-42. DOI:10.1016/j.yrtph.2009.05.018 |
| [2] |
F. Liu, Z. Wu, D. Wang, et al., Colloids Surf. A: Physicochem. Eng. Aspects 490 (2016) 207-214. DOI:10.1016/j.colsurfa.2015.11.053 |
| [3] |
J. Chen, X. Sun, L. Lin, X. Dong, Y. He, Chin. J. Chem. Eng. 25 (2017) 775-781. DOI:10.1016/j.cjche.2016.10.014 |
| [4] |
X. Mei, J. Liu, Z. Guo, et al., J. Hazard. Mater. 363 (2019) 99-108. DOI:10.1016/j.jhazmat.2018.09.072 |
| [5] |
J. Li, Y. Li, Z. Xiong, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 2139-2146. DOI:10.1016/j.cclet.2019.04.057 |
| [6] |
M. Zhu, L. Zhang, S. Liu, et al., Chin. Chem. Lett. 31 (2020) 1961-1965. DOI:10.1016/j.cclet.2020.01.017 |
| [7] |
P. Hu, M. Long, Appl. Catal. B: Environ. 181 (2016) 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| [8] |
M.A. Oturan, J. Peiroten, P. Chartrin, A.J. Acher, Environ. Sci. Technol. 34 (2000) 3474-3479. DOI:10.1021/es990901b |
| [9] |
R.M. Cardoso, R.H.O. Montes, A.P. Lima, et al., Electrochim. Acta 176 (2015) 36-43. DOI:10.1016/j.electacta.2015.06.117 |
| [10] |
D. Méndez, R. Vargas, C. Borrás, et al., Appl. Catal. B: Environ. 166- 167 (2015) 529-534. |
| [11] |
N. Wang, T. Zheng, G. Zhang, P. Wang, J. Environ. Chem. Eng. 4 (2016) 762-787. DOI:10.1016/j.jece.2015.12.016 |
| [12] |
G. Liu, J. Zhou, W. Zhao, Z. Ao, T. An, Chin. Chem. Lett. 31 (2020) 1966-1969. DOI:10.1016/j.cclet.2019.12.023 |
| [13] |
X. Xiong, B. Sun, J. Zhang, N. Gao, J. Shen, J. Li, X. Guan, Water Res. 62 (2014) 53-62. DOI:10.1016/j.watres.2014.05.042 |
| [14] |
M. Nie, C. Yan, M. Li, X. Wang, W. Bi, W. Dong, Chem. Eng. J. 279 (2015) 507-515. DOI:10.1016/j.cej.2015.05.055 |
| [15] |
A. Rastogi, S.R. Al-Abed, D.D. Dionysiou, Appl. Catal. B: Environ. 85 (2009) 171-179. DOI:10.1016/j.apcatb.2008.07.010 |
| [16] |
Y.R. Wang, W. Chu, J. Hazard. Mater. 186 (2011) 1455-1461. |
| [17] |
G. Ayoub, A. Ghauch, Chem. Eng. J. 256 (2014) 280-292. DOI:10.1016/j.cej.2014.07.002 |
| [18] |
M. Yang, S. Zhou, Y. Zheng, K. Zheng, Chem. Indust. Eng. Prog. 31 (2012) 2093-2096. |
| [19] |
Z. Wang, RSC Adv. 7 (2017) 30941-30948. DOI:10.1039/C7RA03872K |
| [20] |
B. Lai, Z. Chen, Y. Zhou, P. Yang, J. Wang, Z. Chen, J. Hazard. Mater. 250- 251 (2013) 220-228. |
| [21] |
H. Zhang, Q. Ji, L. Lai, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 1129-1132. DOI:10.1016/j.cclet.2019.01.025 |
| [22] |
X. Zou, T. Zhou, J. Mao, X. Wu, Chem. Eng. J. 257 (2014) 36-44. DOI:10.1016/j.cej.2014.07.048 |
| [23] |
H. Kusic, I. Peternel, S. Ukic, et al., Chem. Eng. J. 172 (2011) 109-121. |
| [24] |
J. Cao, L. Lai, B. Lai, et al., Chem. Eng. J. 364 (2019) 45-56. DOI:10.1016/j.cej.2019.01.113 |
| [25] |
Y.H. Huang, T.C. Zhang, Water Res. 39 (2015) 1751-1760. |
| [26] |
Y. Nakatsuji, Z. Salehi, Y. Kawase, J. Environ. Manag. 152 (2015) 183-191. DOI:10.1016/j.jenvman.2015.01.012 |
| [27] |
L. Tang, J. Tang, G. Zeng, et al., Appl. Surf. Sci. 333 (2015) 220-228. DOI:10.1016/j.apsusc.2015.02.025 |
| [28] |
D. Zhi, Y. Lin, L. Jiang, et al., J. Environ. Manag. 260 (2020) 110-125. |
| [29] |
M.M. Bello, A.A. Abdul Raman, A. Asghar, Process Saf. Environ. Prot. 126 (2019) 119-140. DOI:10.1016/j.psep.2019.03.028 |
| [30] |
L.W. Matzek, K.E. Carter, Chemosphere 151 (2016) 178-188. DOI:10.1016/j.chemosphere.2016.02.055 |
| [31] |
C. Qi, X. Liu, J. Ma, et al., Chemosphere 151 (2016) 280-288. DOI:10.1016/j.chemosphere.2016.02.089 |
| [32] |
J. Li, Q. Ji, B. Lai, D. Yuan, J. Taiwan Instit. Chem. Eng. 80 (2017) 686-694. DOI:10.1016/j.jtice.2017.09.002 |
| [33] |
G.X. Huang, C.Y. Wang, C.W. Yang, P.C. Guo, H.Q. Yu, Environ. Sci. Technol. 51 (2017) 12611-12618. DOI:10.1021/acs.est.7b03007 |
| [34] |
L. Lai, J. Yan, J. Li, B. Lai, Chem. Eng. J. 343 (2018) 676-688. DOI:10.1016/j.cej.2018.01.035 |
| [35] |
M.S. Dieckmann, K.A. Gray, Water Res. 30 (1996) 1169-1183. DOI:10.1016/0043-1354(95)00240-5 |
| [36] |
Z. Yang, C. Shan, W. Zhang, et al., Water Res. 106 (2016) 461-469. DOI:10.1016/j.watres.2016.10.030 |
| [37] |
M. Zhu, L. Zhang, S. Liu, D. Wang, J. Zou, Chin. Chem. Lett. 31 (2020) 1961-1965. DOI:10.1016/j.cclet.2020.01.017 |
| [38] |
J. Li, Q. Liu, Qq. Ji, B. Lai, Applied. Catal. B: Environ. 200 (2017) 633-646. DOI:10.1016/j.apcatb.2016.07.026 |
| [39] |
Q. Ji, J. Li, Z. Xiong, B. Lai, Chemosphere 172 (2017) 10-20. DOI:10.1016/j.chemosphere.2016.12.128 |
| [40] |
B. Lai, Y. Zhou, H. Qin, et al., Chem. Eng. J. 179 (2012) 1-7. DOI:10.1016/j.cej.2010.12.089 |
| [41] |
M. Omran, T. Fabritius, A.M. Elmahdy, et al., Appl. Surf. Sci. 345 (2015) 127-140. DOI:10.1016/j.apsusc.2015.03.209 |
| [42] |
P. Tang, C. Deng, X. Tang, S. Si, K. Xiao, Chem. Eng. J. 210 (2012) 203-211. DOI:10.1016/j.cej.2012.08.089 |
| [43] |
S.H. Do, Y.J. Kwon, S.J. Bang, S.H. Kong, Chem. Eng. J. 221 (2013) 72-80. DOI:10.1016/j.cej.2013.01.097 |
| [44] |
N.S. Mcintyre, D.G. Zetaruk, Anal. Chem. 49 (1977) 1521-1529. DOI:10.1021/ac50019a016 |
| [45] |
A. Masic, A.T. Santos, B. Etter, K.M. Udert, K. Villez, Water Res. 85 (2015) 244-254. DOI:10.1016/j.watres.2015.08.031 |
| [46] |
R.V. Siriwardane, J.M. Cook, J. Colloid Interf. Sci. 108 (1985) 414-422. DOI:10.1016/0021-9797(85)90280-2 |
| [47] |
H. Cheng, W. Xu, J. Liu, et al., J. Hazard. Mater. 146 (2007) 385-392. DOI:10.1016/j.jhazmat.2006.12.038 |
| [48] |
X. Guo, Z. Yang, H. Liu, et al., Sep.Purif. Technol. 146 (2015) 227-234. DOI:10.1016/j.seppur.2015.03.059 |
| [49] |
X. Wei, N. Gao, C. Li, et al., Chem. Eng. J. 285 (2016) 660-670. DOI:10.1016/j.cej.2015.08.120 |
| [50] |
L. Zhao, Y. Ji, D. Kong, et al., Chem. Eng. J. 303 (2016) 458-466. DOI:10.1016/j.cej.2016.06.016 |
| [51] |
Y. Sun, J. Li, T. Huang, X. Guan, Water Res. 100 (2016) 277-295. DOI:10.1016/j.watres.2016.05.031 |
| [52] |
X. Guo, Z. Yang, H. Dong, et al., Water Res. 88 (2016) 671-680. DOI:10.1016/j.watres.2015.10.045 |
| [53] |
F. Ghanbari, M. Moradi, Chem. Eng. J. 310 (2017) 41-62. DOI:10.1016/j.cej.2016.10.064 |
| [54] |
S. Zhao, H. Ma, M. Wang, et al., J. Hazard. Mater. 180 (2010) 86-90. DOI:10.1016/j.jhazmat.2010.03.076 |