 2021, Vol. 32
2021, Vol. 32 


Intensive interests have been devoted to ion-transport polymer electrolytes because they combine the advantages of solid ionic conductors with the ease processing properties inherent to polymer materials. This feature enables polymer electrolytes to show a wide range of applications in energy storage and conversions such as batteries, supercapacitors, and fuel cells [1-4]. Constructing nanostructured ion transport channels in polymer electrolytes is one of the most effective ways to enhance ion conductivity by offering short ion-transport pathways [5-7]. Among diverse nanostructures, the lamellar morphology with nanoscale interlayer spacing is highly desirable due to the reduced tortuosity of the ion-transport pathways, which can efficiently promote ion conduction [8]. The microphase separation of ionomers with different topological structures, such as block copolymers and graft copolymers, can lead to electrolyte membranes with a lamellar microphase morphology and an increase in the overall conductivity performances [9-12]. For instance, Mezzenga and co-workers constructed lamellar proton transport nanochannels in a diblock copolymer, sulfonated polystyrene-block-polymethylbutylene [9]. The resultant lamellar polymer electrolytes exhibited an improved ion conductivity that was one order of magnitude than the samples with isotropic phase and cylindrical hexagonal phase. Lee et al. realized a high ionic conductivity in the multiblock copolymers of sulfonated poly(arylene sulfide sulfone nitrile), due to the formation of uniform lamellar ion-conducting nanochannels in these polymers [10].
The current strategies to fabricate lamellar structured polymer electrolytes are mainly based on the covalent or non-covalent modification of linear block copolymers. On the other hand, comb-shaped copolymers, which change the topological structure of linear polymers to a branched form through tethering side chains to a polymer backbone, show interesting microphase morphologies and considerable mechanical properties. The unique conformational asymmetry of comb copolymers can facilitate the microphase separation between the backbone and side chains, thus achieving tunable domain morphologies. Theoretical simulations demonstrated that an ordered lamellar phase with multi-oriented domains can be obtained in comb copolymers, by adjusting the grafting density of side chains and the volume ratio of the backbone to side chains [13, 14]. Moreover, experimental studies also confirmed this [15, 16]. For instance, Jannasch et al. obtained lamellar morphology in the sulfonated comb copolymer of poly(phenylene oxide)-graft-poly(4-fluorostyrene) [16]. However, it remains a challenge to construct the large-area ionic nanolayers in comb copolymer electrolytes as ion-conducting channels.
Polyoxometalates (POMs), as a large class of well-defined anionic nanoclusters of metal oxides [17], are widely used as inorganic building blocks to prepare functional hybrid materials [18-29], in particular POM-polymer composite electrolytes [30-38], owing to their high ionic conduction and excellent electrochemical stabilities. Unlike small molecular liquid acids, the characteristic of POMs as a rigid solid acid enables them to improve both the proton conductivity and the mechanical strength of polymer matrices simultaneously, performing as multifunctional proton conductors. Moreover, POMs can also tune the self-assembly behaviors of polymer matrices through electrostatically interacting with the polar groups of polymers. Our group has proposed an electrostatic control strategy of using POMs to induce the formation of nanoscale proton-conducting channels in a series of linear block copolymer matrices, resulting in bicontinuous [39] and inverse hexagonal morphologies [40], which demonstrates the promising role of POMs as morphological modulators in fabricating nanostructure polymer electrolytes.
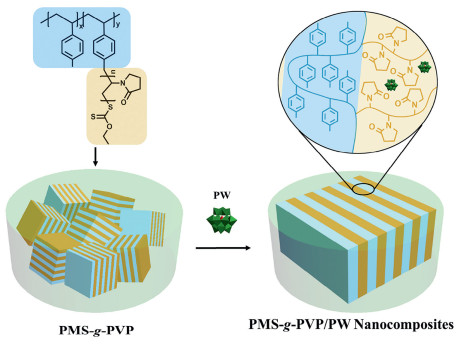
Here, we report the preparation of lamellar structured nanocomposite electrolytes based on POMs and comb copolymers. Poly(4-methlstyrene)-graft-poly(N-vinyl pyrrolidone) (PMS-g-PVP) with the neutral main chains for mechanical support and the polar side chains for ion conduction, was synthesized and electrostatically assembled with a Keggin-type POM, H3PW12O40 (PW), leading to the formation of lamellar nanocomposites with improved long-range regularity in Fig. 1. The morphologies, proton conductivities, and mechanical properties of these nanocomposites were systemically studied. This approach provides a new strategy to fabricate functional comb copolymer electrolytes containing lamellar ion transport nanochannels.

|
Download:
|
| Fig. 1. Schematic illustration of comb copolymer PMS-g-PVP and PMS-g-PVP/PW nanocomposites. | |
{kind=link}
Comb copolymer PMS-g-PVP was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization as shown in Fig. S1 (Supporting information). PMS was selected as the main chain, on which the benzylmethyl groups were brominated and used as the reactive sites for subsequent grafting [41]. Ethylxanthate groups were connected to the backbones of brominated PMS through a nucleophilic substitution reaction to obtain a macromolecular chain transfer agent (PMS-CTA). Then, comb copolymer PMS-g-PVP with a controllable length of PVP side chains was synthesized from the PMS-CTA by grafting monomer, N-vinyl pyrrolidone (NVP), with the grafting density of 0.03. The degree of polymerization (DP) of PVP side chains is controlled to be about 30, according to 1H NMR results. Besides, gel permeation chromatography (GPC) shows that the molecular weight of PVP side chains is 58 kg/mol with a DP value of 29, matching well with the NMR results. The detailed synthesis procedures and characterizations are shown in the Supporting information (Figs. S2–S5 in Supporting information). Such a PVP length gives a PVP volume fraction (fPVP) of 0.42, which can favor the formation of lamellar microphase morphologies.
For the preparation of proton-conducting electrolytes, PW is chosen as the inorganic component, which is deemed as a high proton conductor that provides protons and proton-jumping sites. PW was dissolved with PMS-g-PVP in N,N-dimethylformamide and stirred to gain a homogeneous and transparent solution. In order to facilitate the comparison of the influence of PW content on the nanocomposites, we choose a series of nanocomposite films with increasing PW contents as the research objects. A relatively large interval of 10 wt% in PW content was set for all the samples. Nanocomposite films were obtained after evaporating the solvent with the increasing PW loading of 10 wt%, 20 wt% and 30 wt%, named as NC-1, NC-2 and NC-3, respectively. However, when further loading PW to 40 wt%, the nanocomposite films become very fragile and easy to break into small pieces. Thus, NC-3 is regarded as the optimized sample for further study. Free-standing nanocomposite films of NC-3 are transparent with the introduction of PW, which demonstrates the homogenous dispersion of PW in the PVP domains (Fig. S6 in Supporting information). Furthermore, X-ray diffraction (XRD) experiments showed no characteristic peaks of PW clusters in these films, which further indicated that PW dispersed homogeneously (Fig. S7 in Supporting information). The interaction between PMS-g-PVP and PW was presented by Fourier transform infrared spectroscopy (FTIR). The characteristic vibration bands of the pristine PW with the Keggin unit were observed in the typical FTIR spectra at 1080 cm−1 (P–Oa), 983 cm−1 (W = Od), 892 cm−1 (W–Ob–W) and 806 cm−1 (W–Oc–W), (Oa, the central oxygen atom of PW; Ob, corner-sharing oxygen; Oc, edge-sharing oxygen; Od, terminal oxygen), corresponding to the four kinds of oxygenatoms in PW (Fig. S8 in Supporting information) [34]. As a representation, the FTIR spectrum showed all these vibrations of PW in the NC-3 nanocomposites, which means that PW clusters remain their structural integrity in the nanocomposite films. Meanwhile, an obvious shift was observed in the vibration of W–Oc–W at 817 cm−1 in NC-3 compared with it at 806 cm−1 in PW. The W–Ob–W and W = Od peaks also slightly shifted, appearing at 896 cm−1 and 979 cm−1, respectively. These phenomena of characteristic peak shift indicate that the ambient electrostatic environment of PW changes, that is, PW has an electrostatic interaction with the nitrogen atom in the pyrrolidone unit [40].
Small-angle X-ray scattering (SAXS) is used to study the nanoscale morphologies of the nanocomposite films. The SAXS profiles characterized the low-range scattering peaks in PMS-g-PVP without loading PW. With the increase of PW content, the periodic scattering peaks are clearly observed and are assigned to a lamellar mode (Fig. S9 in Supporting information). In NC-1, NC-2 and NC-3, these scattering peaks are all attributed to the lamellar structure with a long-range order as expected, which indicates that PW plays the role of morphology modulator in inducing a more ordered microphase structure.
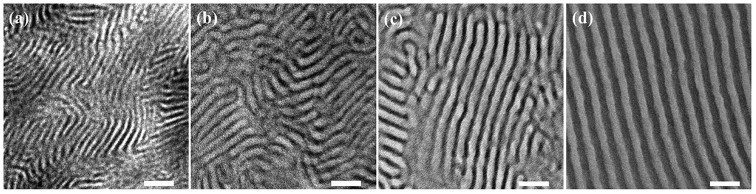
In the PMS-g-PVP/PW nanocomposites, the H atoms of PW can randomly protonate the pyrrolidone groups through electrostaticinteraction, thereby leading to the formation of lamellar nanocomposites with improved long-range regularity. The ultrathin sections of nanocomposite films were prepared for transmission electron microscopy (TEM) measurement to further investigate the microphase separated structures. For the pristine PMS-g-PVP, a short-range, tortuous, and multi-oriented lamellar morphology is observed with light PMS domains and dark PVP domains as shown in Fig. 2a. Note that the PVP domains are stained by iodine to provide a high contrast. As for the films of PW contents of 10 wt% and 20 wt%, the corresponding NC-1 and NC-2 show a lamellar phase with an increased degree of order than the pristine polymer (Figs. 2b and c). As the existence of tungsten element contributing to a high electron density, the dark PVP/PW domains without iodine staining can be clearly recognized compared with the light PMS domains. Further increasing PW loading, the TEM image of NC-3 with a PW loading of 30 wt% exhibits well-defined, long-range regular and highly ordered lamellar morphology in Fig. 2d. The order degree of these lamellar morphologies can be evaluated by comparing the area size of uniformly oriented lamellar domains. The size can be indicated by the diameter of the smallest circle that can cover such a domain, which increases from ~145 nm to ~500 nm with the increased PW content of the samples from 0 to 30 wt% (Fig. S10 and Table S1 in Supporting information). Meanwhile, the distances of the domains also broaden with the increase of PW. The center to center distance between neighbouring PVP domains in NC-3 is about 26.9 nm summed up from TEM images (Fig. S11 and Table S1 in Supporting information). The above results demonstrate that the presence of PW remarkably improves the order degree of the lamellar phase in the PMS-g-PVP matrix and widens the lamellar channels. In our previous works, we found that the electrostatic interaction between POMs and pyrrolidone units in the PVP-b-PS-b-PVP triblock copolymer enabled POMs as morphology modulators to increase the incompatibility between blocks and led to a phase transformation from the ordered structures to inverse hexagonal cylinders [40]. Similarly, in the present work, the improvement from short-range to long-range regularity of lamellar structure can be attributed to the increasing χ value (Flory–Huggins interaction parameter) between PMS and PVP/PW, because the introduction of PW has little effect on the volume fraction of the PVP domains. For instance, the volume fraction only increased by 0.03 even in NC-3 with the 30 wt% PW content.

|
Download:
|
| Fig. 2. TEM images of ultrathin sections of (a) PMS-g-PVP films with iodine staining, (b) NC-1, (c) NC-2 and (d) NC-3 with the scale bar of 50 nm. | |
{kind=link}
In order to further substantiate the composition of PVP/PW domains in NC-3, a high angle annular dark-field scanning transmission electron microscopic (HAADF-STEM) and nanoscale infrared spectroscopy characterization based on atomic force microscopy (AFM-IR) were carried out. In the HAADF-STEM image, PMS domains are darkened because the tungsten components appear brighter against polymers in lamellar structure (Fig. 3a). Then the elemental mapping by energy-dispersive X-ray spectroscopy (EDX) confirmed that the distribution of tungsten and nitrogen, which clearly elucidates that PW clusters and pyrrolidone units interact with each other in the lamellar PVP/PW hybrid domains (Figs. 3b and c). Likewise, from the AFM height images and the related AFM-IR maps that highlight the PVP/PW domains, it can be seen that the domains correspond well with those in the TEM images (Figs. 3d and e). By setting the excitation laser to the 979 cm−1, a clear difference between PMS and PVP/PW can be observed that only PVP/PW domains showed a distinct absorption, which is related to the W = Od absorption band of PW (2D and 3D AFM-IR maps in Figs. 3e and f). The lamellar nanosegregation structures from PVP and PMS components formed in this work also supported the previous simulation and experimental results [13-16], that is, the special molecular structure with almost equal length of side chains in well-defined comb copolymers can facilitate the phase behavior to evolve into a lamellar phase.

|
Download:
|
| Fig. 3. (a) HAADF-STEM image of NC-3. (b) EDX single mapping of W. (c) EDX single mapping of N. (d) AFM height image of NC-3. (e) AFM-IR 2D absorption map at 979 cm−1. (f) AFM-IR 3D absorption map at 979 cm−1. Note that (a–c) correspond to the same region and (d–f) correspond to the same region.Scale bars in (a–c) are 30 nm. Scale bars in (d, e) are 200 nm. | |
{kind=link}
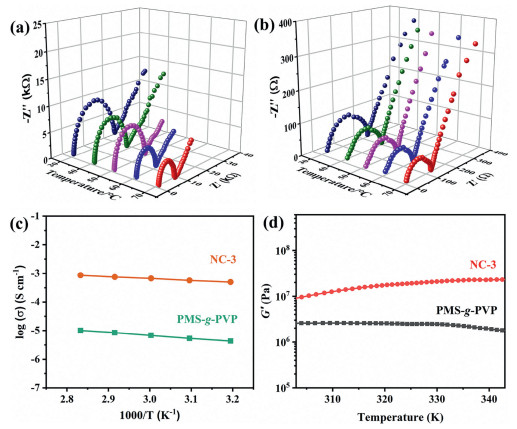
PW can greatly increase the ionic conductivity of PMS-g-PVP, working as proton conductors in the PVP domains. Meanwhile, the neutral PMS main chains provide mechanical support. NC-3 has the highest proton content among all nanocomposites, and thus it is a suitable sample to study the enhanced conductivity of ordered ion channels. The proton conductivities of NC-3 and PMS-g-PVP films in a fully hydrated state were measured from 303 K to 343 K by AC impedance measurements, as shown in the Nyquist plots in Figs. 4a and b (details are shown in Figs. S12, S13 and Table S2 in Supporting information). Note that the films become soft and deformable at higher temperatures and are not suitable for the conductivity test. The conductivity of NC-3 is 4.3 × 10−4 S/cm at 303 K and 7.5 × 10−4 S/cm at 343 K, which is two orders of magnitudes higher than that of pure PMS-g-PVP. The activation energy is relatively low at 0.13 eV by Arrhenius fitting (Fig. 4c), implying that the proton conduction in NC-3 follows the Grotthuss mechanism [42]. Besides, the rigid PW can serve as nano-reinforcers to enhance the mechanical strength of the nanocomposites. The films of pristine PMS-g-PVP and NC-3 were studied by rheometry to assess the mechanical properties of these films during proton conductivity measurements from 303 K to 343 K in Fig. 4d. The storage modulus (G′) of PMS-g-PVP is 2.6 × 106 Pa at 303 K. After loading PW, the G ′ remarkably increases to 1.1 × 107 Pa for NC-3, which reflects that the formation of highly ordered PVP/PW nanolayers can attribute to enhanced mechanical stability.

|
Download:
|
| Fig. 4. Nyquist plots of PMS-g-PVP (a) and NC-3 (b) from 303 K to 343 K. (c) The conductivity of PMS-g-PVP and NC-3 from 303 K to 343 K. (d) Storage modulus G ′ of PMS-g-PVP and NC-3 in the fully hydrated state from 303 K to 343 K. | |
{kind=link}
In summary, we developed an approach to fabricate lamellar-structured nanocomposite polymer electrolytes based on PW and comb copolymer PMS-g-PVP, through the PW-induced phase regulation from a short-range to a long-range ordered lamellar morphology. The electrostatic effect of PW on PVP contributes to the ordered self-assembly of polymer matrices, leading to the formation of a stable lamellar phase. Meanwhile, PW can also serve as high proton conductors and nano-reinforcers, which can address the trade-off between ionic conductivity and mechanical strength, showing a high proton conductivity of 4.3 × 10−4 S/cm and a stable storage modulus at 1.1 × 107 Pa at room temperature. This study provides a new strategy to design functional polymer electrolytes with lamellar ion transport nanochannels.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 22075097), the Program for JLU Science and Technology Innovative Research Team (No. 2017TD-10), and the Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (No. 2020-09).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.051.
| [1] |
D.T. Hallinan, N.P. Balsara, Annu. Rev. Mater. Res. 43 (2013) 503-525. DOI:10.1146/annurev-matsci-071312-121705 |
| [2] |
D.W. Shin, M.D. Guiver, Y.M. Lee, Chem. Rev. 117 (2017) 4759-4805. DOI:10.1021/acs.chemrev.6b00586 |
| [3] |
Q. Zhou, J. Ma, S. Dong, et al., Adv. Mater. 31 (2019) 1902029. DOI:10.1002/adma.201902029 |
| [4] |
B. Zhao, X. Lu, Q. Wang, et al., Chin. Chem. Lett. 31 (2020) 831-835. DOI:10.1016/j.cclet.2019.06.009 |
| [5] |
M.W. Schulze, L.D. McIntosh, M.A. Hillmyer, T.P. Lodge, Nano Lett. 14 (2014) 122-126. DOI:10.1021/nl4034818 |
| [6] |
G. He, Z. Li, J. Zhao, et al., Adv. Mater. 27 (2015) 5280-5295. DOI:10.1002/adma.201501406 |
| [7] |
K.M. Meek, Y.A. Elabd, J. Mater. Chem. A 3 (2015) 24187-24194. DOI:10.1039/C5TA07170D |
| [8] |
E.B. Trigg, K.I. Winey, Mol. Syst. Des. Eng. 4 (2019) 252-262. DOI:10.1039/C8ME00086G |
| [9] |
L. Rubatat, C. Li, H. Dietsch, et al., Macromolecules 41 (2008) 8130-8137. DOI:10.1021/ma801543q |
| [10] |
D.W. Shin, S.Y. Lee, C.H. Lee, et al., Macromolecules 46 (2013) 7797-7804. DOI:10.1021/ma400889t |
| [11] |
C.M. Evans, G.E. Sanoja, B.C. Popere, R.A. Segalman, Macromolecules 49 (2015) 395-404. |
| [12] |
O. Kim, K. Kim, U.H. Choi, M.J. Park, Nat. Commun. 9 (2018) 5029. DOI:10.1038/s41467-018-07503-4 |
| [13] |
L. Zhang, J. Lin, S. Lin, J. Phys. Chem. B 112 (2008) 9720-9728. DOI:10.1021/jp802770z |
| [14] |
B. Husowitz, P.A. Monson, Macromolecules 43 (2010) 9549-9554. DOI:10.1021/ma101441g |
| [15] |
E.M.W. Tsang, Z. Zhao, Z. Shi, J. Am. Chem. Soc. 129 (2007) 15106-15107. DOI:10.1021/ja074765j |
| [16] |
M. Ingratta, E.P. Jutemar, P. Jannasch, Macromolecules 44 (2011) 2074-2083. DOI:10.1021/ma102879w |
| [17] |
D.L. Long, R. Tsunashima, L. Cronin, Chem. Angew., Int. Ed. 49 (2010) 1736-1758. DOI:10.1002/anie.200902483 |
| [18] |
H. Li, W. Qi, W. Li, et al., Adv. Mater. 17 (2005) 2688-2692. DOI:10.1002/adma.200500893 |
| [19] |
H. Li, S. Pang, S. Wu, et al., J. Am. Chem. Soc. 133 (2011) 9423-9429. DOI:10.1021/ja201594k |
| [20] |
J. Tang, C. Ma, X. Li, et al., Macromolecules 48 (2015) 2723-2730. DOI:10.1021/acs.macromol.5b00214 |
| [21] |
S. Chai, T. Xu, X. Cao, et al., Chin. J. Polym. Sci. 37 (2019) 797-805. DOI:10.1007/s10118-019-2262-8 |
| [22] |
S. Chai, X. Cao, F. Xu, et al., ACS Nano 13 (2019) 7135-7145. DOI:10.1021/acsnano.9b02569 |
| [23] |
H. Li, L. Wu, Polym. Int. 69 (2019) 665-667. |
| [24] |
H. Bian, X. Zhang, H. Zhao, N. Zhang, Chin. Chem. Lett. 30 (2019) 1097-1099. DOI:10.1016/j.cclet.2019.01.030 |
| [25] |
L.J. Ren, H.K. Liu, H. Wu, et al., Adv. Mater. 32 (2020) 1805863. DOI:10.1002/adma.201805863 |
| [26] |
J. Xiao, Q. He, S. Qiu, et al., Sci. China Chem. 63 (2020) 792-801. DOI:10.1007/s11426-019-9709-7 |
| [27] |
J. Zhang, H. Wei, J. Tan, et al., Sci. China Chem. 61 (2018) 328-335. DOI:10.1007/s11426-017-9137-7 |
| [28] |
Y. Liao, N. Liu, Q. Zhang, W. Bu, Macromolecules 47 (2014) 7158-7168. DOI:10.1021/ma501343s |
| [29] |
Q. Liu, X. Wang, Matter 2 (2020) 816-841. DOI:10.1016/j.matt.2020.01.020 |
| [30] |
S. Lu, X. Xu, J. Zhang, et al., Adv. Energy Mater. 4 (2014) 1400842. DOI:10.1002/aenm.201400842 |
| [31] |
H. Ma, B. Liu, B. Li, et al., J. Am. Chem. Soc. 138 (2016) 5897-5903. DOI:10.1021/jacs.5b13490 |
| [32] |
Z. Zheng, Q. Zhou, M. Li, P. Yin, Chem. Sci. 10 (2019) 7333-7339. DOI:10.1039/C9SC02779C |
| [33] |
X. Yuan, C. Sun, J. Duan, et al., J. Mater. Chem. A 7 (2019) 15924-15932. DOI:10.1039/C9TA04714J |
| [34] |
L. Zhai, H. Li, Molecules 24 (2019) 3425. DOI:10.3390/molecules24193425 |
| [35] |
X. Liu, Y. Li, J. Xue, et al., Nat. Commun. 10 (2019) 842. DOI:10.1038/s41467-019-08622-2 |
| [36] |
T. Wen, L. Qiu, Z. Zheng, et al., Macromolecules 53 (2020) 1415-1421. DOI:10.1021/acs.macromol.0c00020 |
| [37] |
M. Zhang, A.M. Zhang, Y. Chen, et al., Energy Storage Mater. 29 (2020) 172-181. DOI:10.1016/j.ensm.2020.04.017 |
| [38] |
B. Liu, B. Hu, J. Du, et al., Angew. Chem. Int. Ed. 60 (2021) 6076-6085. DOI:10.1002/anie.202012079 |
| [39] |
L. Zhang, T. Cui, X. Cao, et al., Int. Ed. 56 (2017) 9013-9017. DOI:10.1002/anie.201702785 |
| [40] |
L. Zhai, S. Chai, G. Wang, et al., Macromol. Rapid Commun. 41 (2020) 2000438. DOI:10.1002/marc.202000438 |
| [41] |
H.K. Kim, M. Zhang, X. Yuan, et al., Macromolecules 45 (2012) 2460-2470. DOI:10.1021/ma202492d |
| [42] |
X. Wu, J.J. Hong, W. Shin, et al., Nat. Energy 4 (2019) 123-130. DOI:10.1038/s41560-018-0309-7 |