 2021, Vol. 32
2021, Vol. 32 


Dynamic covalent bonds (DCBs) have attracted considerable attention and have a wide range of applications in dynamic combinatorial chemistry, stimuli-responsive systems and supramolecular chemistry owing to its specific reversible feature that DCBs can be cleaved and reformed under certain conditions [1]. Examples of DCBs including disulfide bonds, imine bonds, boronic ester and so on have successfully provided convenient methods to fabricate various 2D macrocycles [2] and 3D molecular cages [3] by external stimuli. The dynamic bond reaction can be tuned by various changes of reaction conditions such as temperature [4], light [1a, 5], pH [1c, 6] and guest molecules [7]. Compared to other stimuli, light is an outstanding stimulus with features of accurate controllability and friendly environment and has extensively used to manipulate the dynamic process [5a, 8]. Disulfide bond-containing molecules can be triggered by irradiation of UV light to undergo dynamic covalent reaction. Compared with disulfide bonds (~240 kJ/mol), diselenide bonds with lower bond energy (170 kJ/mol) show more robust activity under much milder conditions [1a, 5c, 9]. And it has been demonstrated that metathesis reaction of diselenide bonds can take place by irradiation of visible light and stopped under dark [1a]. Dynamic covalent reaction based on diselenide and S-Se by the irradiation of visible light has been employed to construct polymer, responsive material [10]. However, to the best to our knowledge, the fabrication of macrocycles based on dynamic covalent containing-selenium bonds under irradiation of light is rarely reported [2a].
Macrocycles have extensive applications in many scientific communities. Especially, the discovery of crown ethers plays a seminal role in the development of modern supramolecular chemistry [11]. During these decades, a significant number of artificial macrocyclic structures such as cucurbiturils [12], calixarenes [13], cyclophanes [14] and pillararenes [15] have been developed. Macrocyclic compounds served as a type of crucial supramolecular host and have been extensively used in molecular recognitions [11d, 12c, 15d, 16], molecular machines [17], smart materials [13b, 18] and so on. Motivated by this, the fabrication of new macrocycles with novel structures and functions has become an essential goal in the supramolecular field. The introduction of various functional units or elements into the skeleton of macrocyclic compounds is one important strategy to obtain novel functional macrocycles. However, rational and efficient synthesis methods remain very rare, especially for the preparation of the macrocycle itself containing visible light-sensitive DCB.
In this work, we introduce a diselenide bond to substitute an oxygen atom in traditional oxacrown ether and present the synthesis of this novel diselenide-containing crown ether BC7Se2 using the potassium ion as a template. It is found that BC7Se2 can take place metathesis reaction to form cyclic oligomers (Scheme 1) in solution through the dynamic covalent exchange of intermolecular diselenide bonds under visible light without any other adducts and the concentration of BC7Se2 predominate the size of oligomers in this process. The decomposition reaction of oligomers is controlled by topological structures and solvents. Furthermore, potassium can inhibit the polymerization of BC7Se2 and promote the depolymerization of oligomers.

|
Download:
|
| Scheme 1. Metathesis reaction between monomer BC7Se2 and its oligomers irradiated by visible light. Insert is the structure of PEGSe2 as control. | |
{kind=link}
According to conventional approaches to fabricate oxacrown ether, the cyclization of BC7Se2 was carried out in the presence of potassium cation, which served as a template to induce cyclization reaction shielded from light (Scheme S1 in Supporting information). BC7Se2 was purified by silica chromatography column protection from light and obtained as a yellow solid. Interestingly, the 1H NMR spectrum showed there were more than two components in the CDCl3 solution. As shown in Fig. S12 (Supporting information), impurity signals not only in the aromatic region (7.15, 7.00 and 6.95 ppm) but also in the upfield (3.88–3.85 and 3.75–3.71 ppm), which were adjacent to OCH2 of BC7Se2, were considered to be the oligomers of BC7Se2. The thin layer chromatography also verified BC7Se2 is the predominant component of the isolated product (Fig. S13 in Supporting information). The isolated product was dissolved in refluxing ethyl acetylate and n -heptane was dropwise added. Then the solution was cooled to precipitate relatively pure monomer BC7Se2 shielded from light.
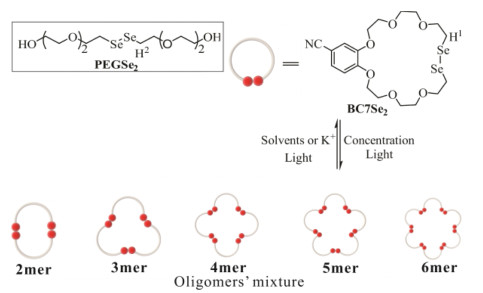
A solution of BC7Se2 with a concentration of 5 mmol/L in CD3CN was prepared and stirred under light generated by two incandescent lamps (220 V, 18 W) at room temperature for 36 h to monitor the dynamic covalent reaction based on BC7Se2. LC–MS spectroscopy was used to determine the structure of the product (Fig. S14a in Supporting information). The sample was diluted to 0.1 mM to characterize by LC–MS coupled with HPLC. There were five molecular ion peaks for dimer of BC7Se2 at m/z 1066.0663 ([M2+NH4]+), 1086.9945 ([M2+K]+), trimer of BC7Se2 at m/z 1589.0842 ([M3+NH4]+), 1610.0133 ([M3+K]+), tetramer of BC7Se2 at m/z 1066.0679 ([M4+2NH4]2+), 1075.5322 ([M4+K+NH4]2+), pentamer of BC7Se2 at m/z 1323.5780 ([M5+2NH4]2+), 1336.5422 ([M5+K+NH4]2+) and hexamer of BC7Se2 at m/z 1588.0867 ([M6+2NH4]2+), 1598.5504 ([M6+K+NH4]2+) (Figs. S14b–g in Supporting information) excepted the ion signal of monomer BC7Se2. The HPLC spectroscopy showed the dimer of BC7Se2 dominated the main component among these oligomers and the content of dimer increased to 4.5% from 1.5% (Fig. S15 in Supporting information). These observations confirmed that diselenide metathesis has occurred. However, it is difficult to distinguish the size of these macrocyclic structures on 1H NMR spectroscopy due to the similar chemical environment of the proton (Figs. S12 and S16 in Supporting information). When the exchange reaction of BC7Se2 at 5 mmol/L was prolonged to 70 h under light, the contents of each macrocyclic molecules determined by LC–MS had no significant increase in HPLC and the reaction reached a balance. Therefore, the results motivated us to explore whether the exchange reaction correlated with the concentration of BC7Se2. Samples with different concentrations of BC7Se2 ranging from 1 mmol/L to 100 mmol/L were prepared and exposed to visible light for 36 h, then were further performed on HPLC to test the dynamic combinatorial library (DCL) components (Fig. 1). The content of each oligomer in DCL was increasing with increasing the original concentration of BC7Se2, and dimer remained kept the highest ratio among oligomers. Take dimer for instance, its content in the DCL was increasing to 15.6%, with the concentration of BC7Se2 increasing to 100 mmol/L. Surprisingly, the new heptamer of BC7Se2 ion peak at m/z 1850.1088 ([M7 + 2NH4]2+) was first found on LC–MS spectroscopy at 100 mmol/L (Fig. S17 in Supporting information). Furthermore, the content distribution of each oligomer in DCL was decreasing, with the macrocyclic structure expanded. Interestingly, no linear oligomers were checked owing to the cyclic structure of the monomer. These observations firmly confirmed that the exchange reaction and the size of oligomers highly depend on the concentration of BC7Se2.

|
Download:
|
| Fig. 1. Metathesis reaction of BC7Se2 irradiated by visible light after 36 h depending on concentration determined by HPLC. The peak for BC7Se2 is omitted for clarity. | |
{kind=link}
A previous study [1a] has confirmed that noncyclic diselenide compounds could undergo diselenide metathesis under visible light and 50% of each reactant took place exchange reaction at equilibrium. However, the exchange reaction of BC7Se2 is weaker than diselenide-containing noncyclic molecules. It is considered that the difference between these two types of structures in dynamic covalent reaction was attributed to the topological structure. So, commercially available BnSe2 was used as a probe to test the exchange reaction between probe and BC7Se2 and noncyclic PEGSe2 that was designed to use as a control compound (Scheme 1), respectively. The metathesis reaction between BnSe2 and PEGSe2 has reached equilibrium under irradiation with visible light over 36 h and about 50% of PEGSe2 reacted with BnSe2 to generate a mixture containing the two reactants and the exchange product in a ratio of 1:1:2 (PEGSe: BnSe: product) determined by H2 integration of PEGSe2 on 1H NMR spectroscopy (Fig. S18 in Supporting information), which is consistent with the literature [1a]. On the contrary, only about 15% of BC7Se2 determined by H1 integration of BC7Se2 on 1H NMR spectroscopy took part in metathesis reaction with BnSe2 to yield a noncyclic structure containing diselenide under the same conditions (Fig. 2). These results suggested that the Se radical generated from BC7Se2 irradiated by visible light preferred to keep cyclic structure owing to the cyclic topology, rather than reacted with Se radical from BnSe. On the other hand, Se radical generated from noncyclic PEGSe2 and BnSe2, respectively, could freely react with each other with the same chance.

|
Download:
|
| Fig. 2. (A) Metathesis reaction between BC7Se2 and BnSe2. (B) Partial 1H NMR spectrum of exchange reaction between BC7Se2 and BnSe2 irradiated by visible light after 36 h, [BC7Se2] = [BnSe2] = 10 mmol/L (400 MHz, CD3CN, 298 K). | |
{kind=link}
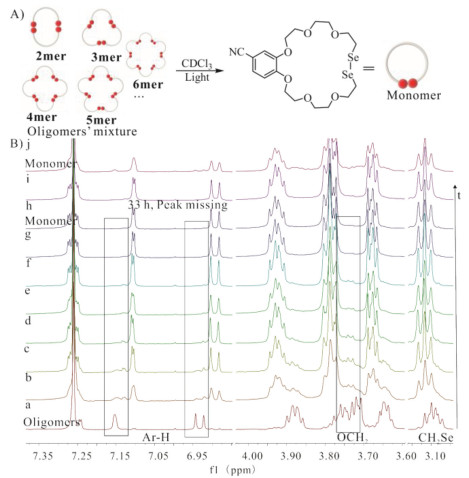
Moreover, the reversible feature of dynamic covalent motivated us to study how to control the cyclic oligomers to depolymerize through dynamic covalent reaction. Solvents have important effects on organic reaction [19]. Thus, solvents effects and guest template were taken into account to investigate the reverse polymerization process of BC7Se2. The mixture with a high content of cyclic oligomers was prepared by concentrating the BC7Se2 solution in dichloromethane by a rotatory evaporator under light due to the polymerization of BC7Se2 was under the control of its concentration. After drying under vacuum, the residue coated on the flask slowly switched from an oily shape to an elastic lump with yellow, which is different from the yellow powder of monomer BC7Se2 (Fig. S20 in Supporting information). A series of the solution containing oligomers of BC7Se2 with a concentration of 5 mmol/L calculated by the molecular weight of BC7Se2 was prepared in CDCl3, CDCl3/CD3CN = 1/2 (v/v), CD3CN, DMSO-d6 respectively under dark, the polarity of which range from low to high. Unfortunately, the oligomers of BC7Se2 could not dissolve in CD3CN. The changes for these four samples under light were monitored by 1H NMR spectroscopy. Before exposed to light, the Ar-H chemical shift of oligomers in the CDCl3 (Fig. 3B–a) was located downfield compared with monomer BC7Se2 (Fig. 3B–j). On the contrary, partial peaks of OCH2 of oligomers mixture shifted upfield compared to monomer. And the signal for the proton of monomer BC7Se2 is very weak in Fig. 3B–a. These observations reconfirmed the polymerization of dynamic covalent reaction for this system is under control of concentration. While the solution of oligomers in CDCl3 was kept under dark for 4 h, there were no significant changes in the1H NMR spectrum (Fig. S19 in Supporting information), however, after exposed exposure to light, oligomers began to depolymerize to form monomer BC7Se2 and took about 33 h to be entirely consumed in CDCl3 (Fig. 3) deduced from it is scarce to observed the 1H NMR peak (in the rectangle) for oligomers (Fig. 3B–i). The results suggested that the depolymerization is dependent on light irradiation. And it took almost 194 h to complete this switch in the mixture solvents (CDCl3/CD3CN = 1/2, v/v) (Fig. S21 in Supporting information). In DMSO- d6 with higher polarity, the content of the oligomers and monomer BC7Se2 seemed to reach a balance, and the depolymerization reaction of oligomers stopped owing to there is no changes on the 1H NMR spectrum after the sample exposed to light for 33 h (Fig. S22 in Supporting information). Although there were no apparent signals for oligomers on its 1H NMR spectrum in CD3CN before exposure to light, which was due to its weak solubility, there were more than two sets of chemical shifts after under light for 8 h (Fig. S23f in Supporting information). One set was attributed to monomer BC7Se2. The other was considered as oligomers with small size due to the weak solubility of oligomers and the decomposition rate become slow after exposure to light for 55 h (Fig. S23i in Supporting information). These phenomena demonstrate solvent effect plays a crucial role in the depolymerization reaction.

|
Download:
|
| Fig. 3. (A) The schematic plot for the decomposition of oligomers. (B) Changes in 1H NMR of oligomers based on BC7Se2 under light. (a) 0 h; (b) 1 h; (c) 2 h; (d) 4 h; (e) 6 h; (f) 8 h; (g) 22 h; (h) 33 h; (i) 55 h; (j) monomer of BC7Se2. [Oligomers] = [BC7Se2] = 5 mmol/L, CDCl3, 400 MHz, 298 K. The peaks in the rectangle represent partial proton of oligomers showed obvious chemical shift changes. | |
{kind=link}
In addition, K+ template effect was used to induce the depolymerization of oligomers in the mixture solvents (CDCl3/CD3CN = 1/2, v/v). In the presence of K+ (3.0 eq.), it is calculated that oligomers took 148 h (Fig. S24k in Supporting information) to depolymerize completely, which is shorter than the case without K+ template (194 h) determined by new peaks of monomer BC7Se2 substituted the signal of oligomers in 1H NMR spectrum (Fig. S21). On the other hand, these two samples were concentrated under light, and the residue was resolved in CDCl3/CD3CN = 1/2 (v/v) under dark to tested by 1H NMR spectroscopy. The spectrum suggested the residue of the sample in the absence of K+ is oligomers (Fig. S25 in Supporting information), and it can repeat depolymerization under light. Interestingly the residue in the presence of K+ still kept monomer form BC7Se2 (Fig. S26 in Supporting information). All these results demonstrated that K+ template could accelerate the decomposition of oligomers based on BC7Se2 and strongly inhibit the polymerization of monomer BC7Se2.
In conclusion, we have described the synthesis of diselenide-containing crown BC7Se2 and its dynamic covalent reaction under the irradiation with visible light. To our surprise, BC7Se2 would rather form oligomers with cyclic structure through dynamic covalent reaction than occurred linear polymerization, and this process was controlled by topological structure and concentration of BC7Se2. However, the depolymerization of oligomers was in the charge of solvents and potassium cation has template effects to enhance this process. Furthermore, the template effect of potassium ion can inhibit monomer BC7Se2 from forming oligomers through metathesis reaction. To date, one method for the decomposition of polymers to reuse monomer under mild conditions is to develop an efficient catalyst, which faced great challenges, otherwise, the development of dynamic covalent offers an opportunity for the design of reusable polymeric monomer [20]. Therefore, the current polymerization and decomposition of BC7Se2 under light would be of particular significance, which may extend the research realm of diselenide bond beyond playing an important role in smart materials.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 21901210, 21901209, 22071196, 22007078), Natural Science Foundation of Shaanxi Province of China (No. 2019JQ-626), Postdoctoral Innovative Talents Supporting Project of China (No. BX20180255), Key R&D Program of Shaanxi Province (Nos. 2019KW-031, 2019KW-038), Fundamental Research Funds for the Central Universities (Nos. 3102019smxy001, 3102018zy051, 3102018jcc007, 3102017OQD045, 3102017OQD040, 3102017OQD115, 3102019ghxm005), State Key Laboratory of Solidification Processing in NPU (No. SKLSP201817). We thank the Analytical & Testing Center of NPU for the characterization of materials. We thank the Analytical & Testing Center of NPU for the characterization of materials.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.043.
| [1] |
(a) S. Ji, W. Cao, Y. Yu, H. Xu, Angew. Chem. Int. Ed. 53 (2014) 6781-6785; (b) P.T. Corbett, J. Leclaire, L. Vial, et al., Chem. Rev. 106 (2006) 3652-3711; (c) M.E. Belowich, J.F. Stoddart, Chem. Soc. Rev. 41 (2012) 2003-2024; (d) M. Mondal, N. Radeva, H. Köster, et al., Angew. Chem. Int. Ed. 53 (2014) 3259-3263. |
| [2] |
(a) J.M.A. Carnall, C.A. Waudby, A.M. Belenguer, et al., Science 327 (2010) 1502-1506; (b) B. Liu, C.G. Pappas, E. Zangrando, et al., J. Am. Chem. Soc. 141 (2019) 1685-1689; (c) S. Ji, H.E. Mard, M. Smet, W. Dehaen, H. Xu, Sci. China Chem. 60 (2017) 1191-1196. |
| [3] |
(a) K. Acharyya, P.S. Mukherjee, Angew. Chem. Int. Ed. 58 (2019) 8640-8653; (b) Y. Jin, C. Yu, R.J. Denman, W. Zhang, Chem. Soc. Rev. 42 (2013) 6634-6654; (c) M.E. Briggs, A.G. Slater, N. Lunt, et al., Chem. Commun. 51 (2015) 17390-17393. |
| [4] |
(a) R.C. Boutelle, B.H. Northrop, J. Org. Chem. 76 (2011) 7994-8002; (b) X. Zheng, Y. Zhang, G. Wu, et al., Chem. Commun. 54 (2018) 3138-3141. |
| [5] |
(a) F. Fan, S. Ji, C. Sun, et al., Angew. Chem. Int. Ed. 57 (2018) 16426-16430; (b) J. Li, J.M.A. Carnall, M.C.A. Stuart, S. Otto, Angew. Chem. Int. Ed. 50 (2011) 8384-8386; (c) B. Rasmussen, A. Sørensen, H. Gotfredsena, M. Pittelkow, Chem. Commun. 50 (2014) 3716-3718. |
| [6] |
(a) Y. Yi, H. Xu, L. Wang, W. Cao, X. Zhang, Chem. Eur. J. 19 (2013) 9506-9510; (b) A. Sanchez-Sanchez, D.A. Fulton, J.A. Pomposo, Chem. Commun. 50 (2014) 1871-1874. |
| [7] |
(a) S. Otto, R.L.E. Furlan, J.K.M. Sanders, Science 297 (2002) 590-593; (b) S. Ito, H. Takata, K. Ono, N. Iwasawa, Angew. Chem. Int. Ed. 52 (2013) 11045-11048; (c) A. Galán, E.C. Esudero-Adán, P. Ballester, Chem. Sci. 8 (2017) 7746-7750; (d) H. Löw, E. Mena-Osteritz, M. von Delius, Chem. Commun. 55 (2019) 11434-11437. |
| [8] |
(a) M.J. Hansen, W.A. Velema, M.M. Lerch, W. Szymanski, B.L. Feringa, Chem. Soc. Rev. 44 (2015) 3358-3377; (b) M. Kathan, S. Hecht, Chem. Soc. Rev. 46 (2017) 5536-5550; (c) H. Frisch, D.E. Marschner, A.S. Goldmann, C. Barner-Kowollik, Angew. Chem. Int. Ed. 57 (2018) 2036-2045; (d) M. Herder, J.M. Lehn, J. Am. Chem. Soc. 140 (2018) 7647-7657; (e) H. Wu, Y. Chen, L. Zhang, et al., J. Am. Chem. Soc. 141 (2019) 1280-1289; (f) Y. Zhou, H.Y. Zhang, Z.Y. Zhang, Y. Liu, J. Am. Chem. Soc. 139 (2017) 7168-7171; (g) H.J. Wang, H.Y. Zhang, H. Wu, et al., Chem. Commun. 55 (2019) 4499-4502; (h) H.G. Fu, H.Y. Zhang, H.Y. Zhag, Y. Liu, Chem. Commun. 55 (2019) 13462-13465. |
| [9] |
(a) N.K. Kildahl, J. Chem. Educ. 72 (1995) 423-424; (b) J. Beld, K.J. Woycechowsky, D. Hilvert, Biochemistry 46 (2007) 5382-5390; (c) J. Beld, K.J. Woycechowsky, D. Hilvert, J. Biotechnol. 150 (2010) 481-489. |
| [10] |
(a) P. Zhao, J. Xia, M. Cao, H. Xu, ACS Macro. Lett. 9 (2020) 163-168; (b) S. Ji, W. Cao, Y. Yu, H. Xu, Adv. Mater. 27 (2015) 7740-7745. |
| [11] |
(a) C.J. Pedersen, J. Am. Chem. Soc. 89 (1967) 2495-2496; (b) Z. Liu, S.K.M. Nalluri, J.F. Stoddart, Chem. Soc. Rev. 46 (2017) 2459-2478; (c) Q. He, G.I. Vargas-Zúñiga, S.H. Kim, S.K. Kim, J.L. Sessler, Chem. Rev. 119 (2019) 9753-9835; (d) X. Yan, D. Xu, X. Chi, et al., Adv. Mater. 24 (2012) 362-369; (e) F. Wang, C. Han, C. He, et al., J. Am. Chem. Soc. 130 (2008) 11254-11255. |
| [12] |
(a) J. Murray, K. Kim, T. Ogoshi, W. Yao, B.C. Gibb, Chem. Soc. Rev. 46 (2017) 2479-2496; (b) S.J. Barrow, S. Kasera, M.J. Rowland, J. del Barrio, O.A. Scherman, Chem. Rev. 115 (2015) 12320-12406. |
| [13] |
(a) R. Kumar, A. Sharma, H. Singh, et al., Chem. Rev. 119 (2019) 9657-9721; (b) D.S. Guo, Y. Liu, Chem. Soc. Rev. 41 (2012) 5907-5921. |
| [14] |
(a) H.Y. Zhou, Q.S. Zong, Y. Han, C.F. Chen, Chem. Commun. 56 (2020) 9916-9936; (b) X. Wang, F. Jia, L.P. Yang, H. Zhou, W. Jiang, Chem. Soc. Rev. 49 (2020) 4176-4188. |
| [15] |
(a) K. Wada, T. Kakuta, T.A. Yamagishi, T. Ogoshi, Chem. Commun. 56 (2020) 4344-4347; (b) H. Zhang, Z. Liu, Y. Zhao, Chem. Soc. Rev. 47 (2018) 5491-5528; (c) T. Ogoshi, T.A. Yamagishi, Y. Nakamoto, Chem. Rev. 116 (2016) 7937-8002. |
| [16] |
(a) S. Peng, Q. He, G.I. Vargas-ZúñIGA, et al., Chem. Soc. Rev. 49 (2020) 865-907; (b) D.H. Qu, Q.C. Wang, Q.W. Zhang, X. Mang, H. Tian, Chem. Rev. 115 (2015) 7543-7588. |
| [17] |
(a) Y. Wu, M. Frasconi, W.G. Liu, et al., J. Am. Chem. Soc. 142 (2020) 11835-11846; (b) S. Erbas-Cakmak, D.A. Leigh, C.T. McTernan, A.L. Nussbaumer, Chem. Rev. 115 (2015) 10081-10206; (c) N.H. Evans, P.D. Beer, Chem. Soc. Rev. 43 (2014) 4658-4683; (d) S. Saha, J.F. Stoddart, Chem. Soc. Rev. 36 (2007) 77-92. |
| [18] |
(a) D. Xia, P. Wang, X. Ji, N, et al., Chem. Rev. 120 (2020) 6070-6123; (b) G. Yu, K. Jie, F. Huang, Chem. Rev. 115 (2015) 7240-7303. |
| [19] |
(a) H. Zheng, H. Ye, X. Yu, L. You, J. Am. Chem. Soc. 114 (2019) 8825-8833; (b) A. Chao, I. Negulescu, D. Zhang, Macromolecules 49 (2016) 6277-6284. |
| [20] |
A. Rahimi, J.M. García, Nat. Rev. Chem. 1 (2017) 0046. |