 2021, Vol. 32
2021, Vol. 32 


b School of Environmental and Chemical Engineering, Foshan University, Foshan 528000, China
As a high chemical stability, cost-effective and non-toxic earth oxide, metal oxide (like TiO2, Fe2O3, WO3, etc.) has been widely used in photocatalytic water splitting field since TiO2 was first used as a photoanode to splitting water into hydrogen and oxygen in 1972 [1-4]. However, metal oxide photocatalysts suffer from a high recombination rate of photoexcited carriers and low light absorption efficiency, which leads to low photocatalytic activity [5, 6]. A feasible solution is to design a novel nanostructure, such as one-dimensionally nanorod arrays (NRAs), owing to its large surface area, high aspect ratio and short diffusion length [7]. These unique properties of NRAs enhance light adsorption in the axial direction and carrier separation in the radial direction [8]. Unfortunately, despite these merits, NRAs still face a serious obstacle: sluggish water oxidation kinetic, which causes a slow on-set potential [9, 10].
Generally, the sluggish water oxidation kinetic will cause slow mass transfer efficiency, especially for photoelectrochemical (PEC) system [11, 12]. At the photoelectrode, there are two kinds of reactions (Fig. S1 in Supporting information): one is the generation and separation of photoexcited carrier including electrons and holes inside the photoelectrode at the depletion layer; The other is the reactant/product-mediated reaction occurs at the interface between the photoelectrode and the electrolyte [13, 14]. The reactant of water molecules or OH− ions need to arrive at the surface of the photoanode and react with the holes to produce radical (like ·OH) or oxygen molecules. Subsequently, the products need to migrate and diffuse to the bulk of the solution [15]. This process is defined as mass transfer for photoelectrode. Especially at large bias or under strong illumination, the reaction occurs very fast. If the migration and diffusion are not fast enough, the mass transfer will inhibit the increase of PEC activity. This problem will be more apparent for NRAs, where the space among the NRAs is related narrow [16]. As a result, enhancing water oxidation kinetic should be considered for designing an effective photoanode.
To overcome this problem, many strategies have been deployed. Such as deposition of water oxidation cocatalysts, doping charge-abundant heteroatoms, decorating with noble metal nanoparticles and constructing heterojunctions [17-19]. For example, integrating the photoanode with oxygen evolution cocatalysts like cobalt-phosphate (Co-Pi) [20-22] and iridium oxide (IrO2) [23-25] can promote the water oxidation kinetic, but the interface between the main photoanode and the cocatalysts need to be close to ensure effective charge transfer. Besides, doping is a commonly used strategy to tune the electrical properties of semiconductors, but the doping foreign atoms would inevitably introduce some defects, which act as carrier traps and recombination centers during water oxidation [26, 27]. Thus, the construction heterojunctions in the meantime is an effective method to solve the problem [28, 29]. However, how to design metal oxide-based heterojunction system with good water oxidation kinetic efficiency is a great challenge. Overall, it is highly desirable to develop photoanodes with highly efficient water oxidation kinetic and mass transfer efficiency [30-32].
Herein, we introduce a strategy to improve the mass transfer by enhancing the water oxidation kinetic. We prepare very small NiO clusters with Ni3+ states on the surface by using a hydrothermal method with Nickel acetylacetonate as Ni source. The NiO cluster can be decorated on TiO2 NRAs (TiO2@NiO NRAs). The main step of this fabrication is illustrated in Fig. 1a. Ni3+ state can greatly promote the water oxidation kinetic on the surface. TiO2 NRAs without NiO clusters exhibit a very slow increase of photocurrent density at related bias of 0.7 V vs. RHE when the illumination intensity increases. At larger bias (1.6 V vs. RHE), the increase of photocurrent density becomes slower and slower with stronger illumination intensity. These results indicate that TiO2 NRAs suffer from serious mass transfer inhibition, which should be due to its sluggish water oxidation kinetic. With the decoration of NiO clusters, the photocurrent density increases proportionally with increasing illumination, indicating good mass transfer with the help of water oxidation cocatalyst of NiO clusters. The enhancement of water oxidation kinetic by NiO clusters is confirmed by a series of electrochemical measurements. Also, the photocurrent density of TiO2@NiO NRAs (1.1 mA/cm2) is superior to that of the pristine TiO2 NRAs (0.58 mA/cm2) with an on-set potential shifted negatively by 0.18 V vs. RHE.

|
Download:
|
| Fig. 1. (a) Schematic illustration for the fabrication of TiO2 NRAs and TiO2@NiO NRAs. (b) SEM image and (c) TEM image of TiO2 NRAs. (d) TEM image and (e) HRTEM image of TiO2@NiO NRAs. | |
{kind=link}
The preparation process of TiO2@NiO NRAs was schematically described in Fig. 1a (more details about the experiment and XRD analysis are provided in Section 1 and 2 in Supporting information). TiO2 NRAs were firstly synthesized on an FTO glass by a simple hydrothermal method. Upon the completion of the reaction, a white homogeneous film of vertically aligned nanorods was observed on the FTO substrate (Fig. 1b). The nanorod exhibits a diameter of about 100 nm (Fig. 1c). Subsequently, NiO clusters were growing on the surface of TiO2 NRAs in a mixture solution of nickel acetylacetonate and tert-butanol solvent. It can be seen that some NiO clusters are immobilized uniformly and intimately on the surface of TiO2 NRAs but the structure of nanorod remains after the hydrothermal reaction (Fig. 1d). The NiO cluster is about 25 nm in diameter and the single NiO nanoparticle is about 4 nm. Fig. 1e exhibits the high-resolution transmission electron microscopy (HRTEM) image of TiO2@NiO NRAs. The lattice fringe spacing of 0.32 nm matches well with the (110) plane of TiO2. While the lattice fringe with the inter-planar distance of 0.24 nm, which corresponds to the (101) plane of NiO. These results indicate that the co-existence of TiO2 and NiO, and the heterostructured TiO2@NiO NRAs were formed. Besides, energy dispersive spectroscopy (EDS) displays the elemental analysis (Fig. S3 in Supporting information). Obviously, the TiO2 NRAs are only comprised of Ti and O, and the TiO2@NiO NRAs is only consisted of Ti, O and Ni, suggesting that the successful fabrication of purity TiO2 NRAs and TiO2@NiO NRAs.
To evaluate the chemical composition and oxide state of the TiO2@NiO NRAs, XPS measurements were conducted on the TiO2@NiO NRAs. Fig. S4 (Supporting information) shows the high-resolution XPS spectrum of Ti 2p peak of the TiO2@NiO NRAs. The main two peaks at 464.2 eV and 458.3 eV, which corresponds to the Ti 2p1/2 and Ti 2p3/2, respectively [33]. This result indicates a normal Ti4+ oxidation state, which in accordance with the literature reported previously [34]. In the Ni 2p XPS spectrum (Fig. 2a), the peak at 856.3 eV can be attributed to Ni3+ (NiOOH or Ni2O3) in the Ni 2p fine spectrum [35]. Peak at 862.2 eV is its satellite peak. This is in good agreement with a previous report [35]. It is more reasonable to attribute the Ni3+ as Nickle oxy-hydroxide (NiOOH) because Ni2O3 is not as stable at low temperatures in the presence of water [36]. This suggests there is a layer of NiOOH on the surface of the NiO. This layer is possibly formed during the hydrothermal reaction. This indicates that the surface of NiO clusters is rich in Ni3+ states. Ni3+ states are highly active for water oxidation [37]. Therefore, one advantage of the NiO cluster is the presence of plenty of Ni(III) states under ambient conditions. In the O 1s spectrum (Fig. 2b), the peak can be deconvoluted into three peaks centered at 529.4 eV, 530.9 eV and 532.1 eV, correlating to lattice oxygen, hydroxyl groups and adsorbed water, respectively [38].

|
Download:
|
| Fig. 2. High-resolution XPS spectrum of (a) Ni 2p and (b) O 1s of TiO2@NiO NRAs. (c) Valance XPS spectrum of TiO2@NiO NRAs. (d) Energy diagram of TiO2@NiO NRAs. | |
{kind=link}
In the valance XPS spectrum, the maximum energy often reflects the distance between the valance band and the Fermi level [39]. In Fig. 2c, the spectrum displays two appreciable maximum energies at 1.1 eV and 0.4 eV. The former should be attributed to TiO2, which is close to the reported value of 1.2 eV for TiO2 powders [40]. The small difference may be due to morphology differences. The latter is possibly related to p-type NiO because the valance of near the Fermi level. Thus, the band structure of the TiO2@NiO NRAs can be obtained (Fig. 2d). Such a band structure is beneficial to charge separation and transfer. Due to photoexcited holes in the TiO2 NRAs will preferentially transfer to the NiO clusters with lower valance band. Then those holes generated in the TiO2 NRAs are easy to accumulate on the NiO clusters to participate in the oxidation reaction.
The on-set potential is one of the most important metrics for the photocurrent curves (more details see Section 3 in Supporting information) [41]. It is considered as the potential where PECs begin to generate photocurrent noticeably, and should be as cathodic as possible. In an ideal case, the on-set potential should be very close to the flat band potential. However, it is difficult to reach the flat band potential because of poor charge transfer and slow water oxidation kinetics. The on-set potential of TiO2 NRAs (0.64 V vs. RHE) is 0.72 V positive than its flat band potential measured in the Mott-Schottky plot (-0.68 V vs. Ag/AgCl, -0.08 V vs. RHE) (Fig. S5 in Supporting information). This is partially due to slow water oxidation kinetics. Fig. 3a shows the current density versus potential in the dark, it can be seen that TiO2 NRAs begins to oxidize water at 2.15 V vs. RHE, which is more positive than the theoretical value of 1.23 V. This confirms that TiO2 NRAs suffer from sluggish water oxidation kinetic. According to the literature, the onset potential is defined as the potential where the rise of photocurrent exceeds 0.2 mA cm−2 V-1 in the first-order derivative of photocurrent against the potential [42]. It is clear that the onset potential of TiO2@NiO is at 1.62 V vs. RHE, which is much earlier than that of pure TiO2 at 2.15 V vs. RHE (Fig. S6 in Supporting information). Besides, the water oxidation current density of TiO2@NiO (1.4 μA/cm2) is much larger than that of pure TiO2 (0.05 μA/cm2) at 2.5 V vs. RHE. This all indicates that the NiO cluster with Ni3+ state on the surface facilitates the water oxidation kinetic of the TiO2 NRAs.

|
Download:
|
| Fig. 3. Voltammograms of TiO2 NRAs and TiO2@NiO NRAs (a) in the dark and (b) under AM 1.5 G simulated sunlight (100 mW/cm2) in 0.1 mol/L Na2SO4. (c) The zoom-in view of the green frame in (b). (d) The first-order derivative of the photocurrent density as a function of potential. Light intensity dependent photocurrent on bare TiO2 NRAs and TiO2@NiO NRAs at (e) 0.7 V and (f) 1.4 V vs. RHE. | |
{kind=link}
Figs. 3b and c show the current density versus potential under simulated sunlight irradiation. The on-set potential under illumination is defined as the potential where the change in photocurrent as a function potential exceeds 0.2 mA cm−2 V-1 [42]. As shown in Fig. 3d, the pristine TiO2 NRAs shows a late on-set potential (0.64 V vs. RHE), while, the on-set potential of TiO2@NiO NRAs shift negatively by 0.18 V to 0.46 V vs. RHE, which reduces the external power to drive the PEC process. Besides, the photocurrent increases much faster to a higher level of saturated photocurrent density (from 0.58 mA/cm2 to 1.1 mA/cm2 at 2.5 V vs. RHE). The decreased on-set potential of TiO2@NiO should be attributed to the improvement of water oxidation kinetic.
In this work, we discover that the enhancement of water oxidation kinetic strongly relates to the mass transfer efficiency. Generally, the PEC performances are expected to produce higher photocurrent density with larger bias and stronger illumination, due to more photoexcited electron/hole pairs will be generated upon stronger illumination. The increase of PEC performance with larger bias or stronger illumination is an important parameter to evaluate the activity of the photoelectrode. The photocurrent of TiO2 NRAs as a function of light density is displayed in Figs. 3e and f. At small bias (0.7 V, Fig. 3e), the photocurrent of TiO2 NRAs hardly increases with enhanced illumination. At larger bias (1.4 V, Fig. 3f), the photocurrent increase but deviate from linearity. The increase of illumination does not lead to a proportional increase in photocurrent density. Ideally, all of the increased carriers will contribute to a proportional increase of photocurrent. In our experiment, due to slow water oxidation kinetics, the increased holes are not able to transfer time from the photoanode to the electrolyte, thereby resulting in more serious recombination and slower increase of photocurrent. This is adverse to the PEC system because of the lower utilization of solar energy. By contrast, in the presence of NiO cluster water oxidation cocatalyst, the photocurrent increase linearly with increasing illumination at a small bias of 0.7 V vs. RHE. Importantly, it perverse the linearity even at a large bias (1.6 V vs. RHE). With the aid of NiO clusters, the water oxidation kinetic is greatly promoted and the mass transfer will not inhibit at the PEC system. This suggests the NiO clusters facilitate the mass transfer on TiO2 NRAs by enhancing the water oxidation kinetic, and enhance the utilization efficiency of solar energy in the PEC performances.
As presented in Fig. 4a, similar to the CV curve, the surface charge transfer efficiency (ηtrans) rise quickly from 0.5 V to 1.0 V due to increased external bias, and saturate at large bias (more details see Section 4 in Supporting information). Due to slow water oxidation kinetics, the ηtrans of bare TiO2 NRAs is only 65% at 1.23 V vs. RHE, while that of NiO cluster loaded reach a light level of 90%. This confirms the NiO clusters as highly efficient cocatalyst for water oxidation.

|
Download:
|
| Fig. 4. (a) Surface charge transfer efficiency of TiO2 NRAs and TiO2@NiO NRAs. Time dependence of open-circuit potential of (b) TiO2 NRAs and (c) TiO2@NiO NRAs. (d) Applied bias photon-to-current efficiency (ABPE) of water splitting on TiO2 NRAs and TiO2@NiO NRAs. | |
{kind=link}
In addition, the enhancement of water oxidation kinetic and mass transfer efficiency can also be confirmed by measuring open circuit potential (Voc) (more details see Section 5 in Supporting information). Voc in the dark is determined by the electronic property, carrier density and surface state of the semiconductors [43]. As shown in Figs. 4b and c, the addition of NiO cluster increases the photovoltage (Vph) by 0.07 V, suggesting higher carrier density (nc). This result can be attributed to the faster charge and mass transfer and faster water oxidation kinetic on the NiO-covered surface. Photoexcited holes migrate from the photoanode body to the electrolyte more easily and diminish the possibility of recombination with electrons, then increasing the carrier density in the photoanode (Details see Section 6 in Supporting information).
In general, an external bias is required to activate and promote a PEC. From this viewpoint, a PEC is driven by both external bias and light. Higher bias will boost the solar energy conversion efficiency, but will also lead to direct electrolysis. It is very important to evaluate the contribution of solar energy without considering that of the external bias. The applied bias photon-to-current efficiency (ABPE) can be calculated according to the equation bellows:
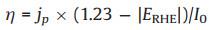
|
(1) |
where η is the efficiency of PEC water splitting, jp is the photocurrent density at the measured potential, I0 is the power density of incident light (100 mW/cm2), and ERHE is the bias potential vs. RHE.
As shown in Fig. 4d, it is obvious that the ABPE of TiO2@NiO NRAs covers a larger potential window than the bare TiO2 NRAs. This is consistent with its earlier on-set potential. Also, the ABPE of TiO2@NiO NRAs is about 3.3 times higher than bare TiO2 NRAs, and it reaches the climax (at 0.89 V) about 0.04 V earlier than the bare TiO2 NRAs. Such a higher ABPE and earlier peak climax indicate that NiO cluster greatly improves the photoconversion efficiency of the TiO2 NRAs.
Besides, to analysis the ABPE after 1.23 V vs. RHE, we also use the following equation bellows to calculate the ABPE:
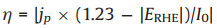
|
(2) |
The negative value after 1.23 V vs. RHE can also be considered and the result is shown in Fig. S7a (Supporting information). It is obvious that the ABPE of TiO2@NiO is higher than TiO2 even in large bias.
In addition, as the water oxidation potential in our experiment is 2.15 V vs. RHE, we also attempt to use 2.15 V vs. RHE as the water oxidation potential to calculate the ABPE (Eq. 3). The calculated result is shown in Fig. S7b (Supporting information). It can be found that the efficiency of TiO2@NiO is also higher than TiO2 at large bias.
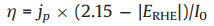
|
(3) |
Except for TiO2, other frequently-used semiconductors, such as WO3 and Fe2O3 have been also investigated as a supporter of cocatalyst NiO, due to their low-cost and earth-abundant. Figs. S8 and S9 (Supporting information) show the current density versus potential under WO3@NiO and Fe2O3@NiO, respectively. The photocurrent can be greatly improved after loading of the cocatalyst NiO in both dark and simulated sunlight. It indicates that NiO can enhance the PEC performance of WO3 and Fe2O3. In addition, ABPE measurement provides another way to illustrate the enhanced PEC activity. As displayed in Fig. S10 (Supporting information), after loading of NiO, the ABPE of WO3@NiO is about 2.9 times higher than bare WO3, and it reaches the climax (at 1.03 V) about 0.09 V earlier than the bare WO3. For Fe2O3, after loading of NiO, the ABPE of Fe2O3@NiO is about 3.5 times higher than bare Fe2O3, and it reaches the climax (at 0.98 V) about 0.12 V earlier than the bare Fe2O3 (Fig. S11 in Supporting information). This result indicates loading of NiO can not only enhance the photoconversion efficiency, but also reduce the applied potential.
In summary, we develop a novel strategy to improve the mass transfer of NRAs photoanode by using Ni-based water oxidation cocatalysts. The TiO2 NRAs suffer from low mass transfer efficiency and sluggish water oxidation kinetic. The photocurrent increase is inhibited, especially at large bias and strong illumination. However, after loading of NiO cluster, the TiO2@NiO NRAs exhibits enhanced photocatalytic activity with faster mass transfer efficiency, larger carrier density, lower on-set potential and better photoconversion efficiency. This work is helpful to understand the PEC performance of NRAs from the viewpoint of mass transfer and deepen our understanding of PEC process. The preparation of Ni-based cocatalyst with Ni3+ is applicable to many metal oxide photoanode, suggesting that this method is widely-acceptable.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the Guangdong Basic and Applied Basic Research Foundation (No. 2019B1515120058), the National Natural Science Foundation of China (No. 51902357), the Natural Science Foundation of Guangdong Province, China (No. 2019A1515012143), the Start-up Funds for High-Level Talents of Sun Yat-sen University (No. 38000-18841209) and the Fundamental Research Funds for the Central Universities (No. 19lgpy153).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.039.
| [1] |
A. Fujishima, K. Honda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [2] |
T.W. Kim, K.S. Choi, Science 343 (2014) 990-994. DOI:10.1126/science.1246913 |
| [3] |
Z. Hu, Z. Shen, J.C. Yu, F. Cheng, Appl. Catal. B 203 (2017) 829-838. DOI:10.1016/j.apcatb.2016.10.079 |
| [4] |
F. Ronconi, Z. Syrgiannis, A. Bonasera, et al., J. Am. Chem. Soc. 137 (2015) 4630-4633. DOI:10.1021/jacs.5b01519 |
| [5] |
Z. Hu, G. Liu, X. Chen, Z. Shen, J.C. Yu, Adv. Funct. Mater. 26 (2016) 4445-4455. DOI:10.1002/adfm.201600239 |
| [6] |
N.C. Zheng, T. Ouyang, Y. Chen, et al., Catal. Sci. Technol. 9 (2019) 1357-1364. DOI:10.1039/C8CY02206B |
| [7] |
T. Zhu, Y. Liang, Y. Wang, et al., Appl. Catal. B 271 (2020) 118945. DOI:10.1016/j.apcatb.2020.118945 |
| [8] |
X. Wang, C. Liow, D. Qi, et al., Adv. Mater. 26 (2014) 3506-3512. DOI:10.1002/adma.201306201 |
| [9] |
Z. Hu, Z. Shen, J.C. Yu, Chem. Mater. 28 (2016) 564-572. DOI:10.1021/acs.chemmater.5b04058 |
| [10] |
P. Kuang, L. Zhang, B. Cheng, J. Yu, Appl. Catal. B 218 (2017) 570-580. DOI:10.1016/j.apcatb.2017.07.002 |
| [11] |
Z. Gu, X. An, H. Lan, et al., Appl. Catal. B 244 (2019) 740-747. DOI:10.1016/j.apcatb.2018.12.009 |
| [12] |
M.I. Jaramillo-Gutiérrez, M.I. Carreño-Lizcano, J.O. Ruiz-Lizarazo, et al., Chem. Eng. J. 386 (2020) 123895. DOI:10.1016/j.cej.2019.123895 |
| [13] |
D. Li, K. Xiong, Z. Yang, et al., Today 175 (2011) 322-327. DOI:10.1016/j.cattod.2011.04.007 |
| [14] |
H. Ren, T. Dittrich, H. Ma, et al., Adv. Mater. 31 (2019) 1807204. DOI:10.1002/adma.201807204 |
| [15] |
A. Liao, R. Chen, F. Fan, et al., Chem. Commun. 54 (2018) 13817-13820. DOI:10.1039/C8CC08350A |
| [16] |
T. Zhou, J. Wang, S. Chen, et al., Appl. Catal. B 267 (2020) 118599. DOI:10.1016/j.apcatb.2020.118599 |
| [17] |
H. Wang, X. He, W. Li, et al., Chem. Commun. 55 (2019) 11382-11385. DOI:10.1039/C9CC05331J |
| [18] |
Y.S. Chen, L.Y. Lin, J. Power. Sources 436 (2019) 226842. DOI:10.1016/j.jpowsour.2019.226842 |
| [19] |
Y. Pi, B. Liu, Z. Li, et al., J. Colliod. Interface Sci. 545 (2019) 282-288. DOI:10.1016/j.jcis.2019.03.041 |
| [20] |
G.M. Carroll, D.K. Zhong, D.R. Gamelin, Energy Environ. Sci. 8 (2015) 577-584. DOI:10.1039/C4EE02869D |
| [21] |
D.K. Zhong, D.R. Gamelin, J. Am. Chem. Soc. 132 (2010) 4202-4207. DOI:10.1021/ja908730h |
| [22] |
E.S. Kim, N. Nishimura, G. Magesh, et al., J. Am. Chem. Soc. 135 (2013) 5375-5383. DOI:10.1021/ja308723w |
| [23] |
F.A. Frame, T.K. Townsend, R.L. Chamousis, et al., J. Am. Chem. Soc. 133 (2011) 7264-7267. DOI:10.1021/ja200144w |
| [24] |
M. Higashi, K. Domen, R. Abe, Energy Environ. Sci. 4 (2011) 4138-4147. DOI:10.1039/c1ee01878g |
| [25] |
X. Liu, Y. Li, S. Peng, et al., Photochem. Photobiol. Sci. 12 (2013) 1903-1910. DOI:10.1039/c3pp50167a |
| [26] |
J. Zhou, L. Yu, Q. Zhu, C. Huang, Y. Yu, J. Mater. Chem. A 7 (2019) 18118-18125. DOI:10.1039/C9TA06347A |
| [27] |
B. Zhang, H. Zhang, Z. Wang, et al., Appl. Catal. B 211 (2017) 258-265. DOI:10.1016/j.apcatb.2017.03.078 |
| [28] |
Y. Hou, X. Zhuang, X. Feng, Small Methods 1 (2017) 1700090. DOI:10.1002/smtd.201700090 |
| [29] |
Y. Hou, M. Qiu, T. Zhang, et al., Adv. Mater. 29 (2017) 1701589. DOI:10.1002/adma.201701589 |
| [30] |
Y. Hou, M. Qiu, T. Zhang, et al., Adv. Mater. 29 (2017) 1604480. DOI:10.1002/adma.201604480 |
| [31] |
Y. Hou, M. Qiu, G. Nam, et al., Nano Lett. 17 (2017) 4202-4209. DOI:10.1021/acs.nanolett.7b01030 |
| [32] |
J. Ke, M.A. Younis, Y. Kong, et al., Nano-Micro Lett. 69 (2018) 1-27. |
| [33] |
Z. Liang, H. Hou, Z. Fang, et al., ACS Appl. Mater. Interfaces 11 (2019) 19167-19175. DOI:10.1021/acsami.9b04059 |
| [34] |
Y. Pang, Y. Li, G. Xu, et al., Appl. Catal. B 248 (2019) 255-263. DOI:10.1016/j.apcatb.2019.01.077 |
| [35] |
X. Wang, K. Cheng, S. Dou, et al., Appl. Surf. Sci. 467 (2019) 136-143. |
| [36] |
Z. Tan, P. Liu, H. Zhang, et al., Chem. Commun. 51 (2015) 5695-5697. DOI:10.1039/C5CC00661A |
| [37] |
K. Fominykh, J.M. Feckl, J. Sicklinger, et al., Adv. Funct. Mater. 24 (2014) 3123-3129. DOI:10.1002/adfm.201303600 |
| [38] |
B.S. Yeo, A.T. Bell, J. Phys. Chem. C 116 (2012) 8394-8400. |
| [39] |
N. Li, K. Du, G. Liu, et al., J. Mater. Chem. A 1 (2013) 1536-1539. DOI:10.1039/C2TA01012G |
| [40] |
X.B. Chen, L. Liu, P.Y. Yu, S.S. Mao, Science 331 (2011) 746-750. DOI:10.1126/science.1200448 |
| [41] |
S.D. Tilley, M. Cornuz, K. Sivula, M. Gratzel, Angew. Chem. Int. Ed. 49 (2010) 6405-6408. DOI:10.1002/anie.201003110 |
| [42] |
C. Du, X. Yang, M.T. Mayer, et al., Angew. Chem. Int. Ed. 52 (2013) 12692-12695. DOI:10.1002/anie.201306263 |
| [43] |
G. Liu, J. Shi, F. Zhang, et al., Angew. Chem. Int. Ed. 53 (2014) 7295-7299. DOI:10.1002/anie.201404697 |