 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Macromolecular Synthesis and Functionalization of Ministry of Education, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China;
c State Key Laboratory of Nuclear Physics and Technology, School of Physics, Peking University, Beijing 100871, China;
d Department of Chemical & Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore
Volatile molecules such as fragrances are widely used in cosmetics, household products, medicine, and other industries [1]. They cannot only affect our emotions [2, 3], but also have anti-inflammatory, antibacterial, and antioxidant effects [1]. Fragrances are usually volatile [4], which dramatically limits their industrial applications. The fragrance concentrations in the air should be maintained at a certain level for maximized effect, but the volatile nature makes it vary with temperature and season. Therefore, it is necessary to effectively encapsulate fragrant molecules and tune their release rate.
Various materials have been developed for fragrance encapsulation, including polymeric nanoparticles [5, 6], zeolite [7, 8] and metal-organic framework (MOF) [9, 10]. Among these materials, MOFs, formed by coordination of metal ions and organic ligands, have several advantages such as stable structure, permanent porosity, large specific surface area and adjustable pore size. MOFs have been widely used in catalytic [11, 12], adsorption and separation [13, 14], sensors [15, 16] and drug delivery [17-19]. The porous structures of MOFs can be functionalized with various groups, and the pore sizes can be adjusted with ligands of different lengths [20, 21]. Rational design of pore chemistry (postsynthetic modification) endows MOF materials with high absorption capacity and selectivity to specific gas molecules through the controlling over the intermolecular host-guest interaction [20]. Postsynthetic modification can also enhance the stability of MOF [22, 23]. Recently, we have applied MOFs for encapsulation and controlled release of volatile molecules, e.g., fragrance. We found that the polarity of the MOF pore affects the release of encapsulated molecules. The incorporation of polar groups such as hydroxyl groups into the structures of MOFs can sustain the release of polar fragrances due to the formation of hydrogen bond interactions [9, 10].
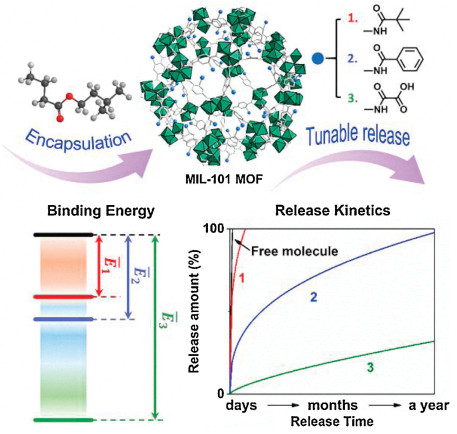
One of the most effective approaches to regulate the loading capacity and release profile of guest molecules in a porous matrix is to change the pore surface chemistry, thereby affecting the host-guest interactions [9, 17, 24]. In this work, we hypothesize that tailoring the pore chemistry of MOFs may enable the tunable release of volatile molecules from MOF materials (Scheme 1). The pore surface functionalization with different organic groups can vary the pore size, polarity and hydrophobicity, thus changing the host-guest interactions and controlling over the release of guest molecules. To demonstrate the concept, we chose amine-functionalized and chromium(III)-based MIL-101(Cr)-NH2 MOF, because of the easiness for synthesis and surface modification, the mesoporous pores large enough for loading volatile molecules, and the high chemical, moisture and thermal stability [18, 21, 25, 26]. Postsynthetic modification was used for the chemical modification of the pore surface of MIL-101(Cr)-NH2 through amidation reaction [27]. Fragrances as a typical type of volatile molecules was selected as representative guest molecules.

|
Download:
|
| Scheme 1. Illustration of tuning the release of volatile molecules by adjusting the binding energy between guest molecules and MOFs. | |
{kind=link}
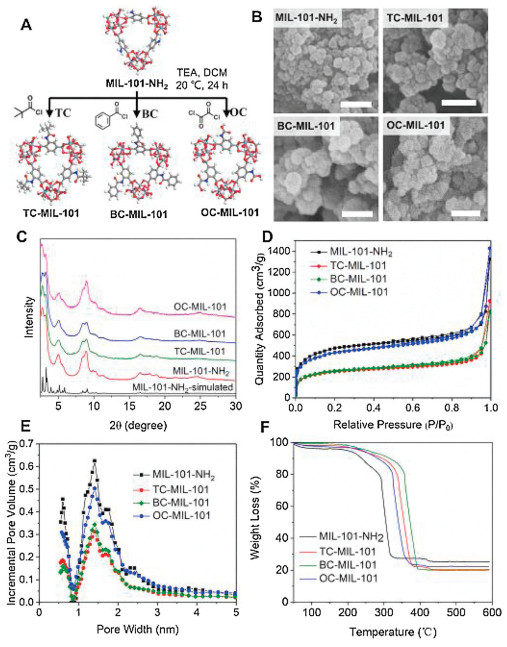
By postsynthetic modification of activated MIL-101-NH2, we obtained four MIL-101 MOFs with different pore surfaces (Fig. 1A), including amine, oxalic acid, benzyl group or tert-butyl group to evaluate their effects on the release of guest molecules. These groups are different in size, polarity and hydrophobicity, and thus may have different interactions with guest molecules. For instance, the previous studies showed that covalent attachment of oxalic acid enhances selectivity of CO2/N2 [28], and the introduction of benzyl group increases the hydrogen uptake capacity [29]. We first synthesized and characterized all the MIL-101 MOFs (MIL-101-NH2, TC-MIL-101, BC-MIL-101 and OC-MIL-101). The scanning electron microscope (SEM) images (Fig. 1B) show that all the MIL-101 MOFs exhibit similar aggregated morphologies composed of spherical or octahedral particles with diameters of around tens of nanometers, similar to previous reports [30]. The powder X-ray diffraction (PXRD) pattern of MIL-101-NH2 is similar to the simulated-MIL-101 and the reported pattern [31, 32] (Fig. 1C). The three modified MOFs possess the same crystalline reflections as initial MIL-101-NH2. These results suggest that the structural topology of the MOFs maintained after postsynthetic modification. The Brunauer Emmett Teller (BET) surface area of MIL-101-NH2, TC-MIL-101, BC-MIL-101 and OC-MIL-101 were calculated to be 1683, 913, 936 and 1562 m2/g, respectively, based on the 77 K N2 sorption isotherms (Fig. 1D). The pore-size distribution curves show that all the crystals have two types of micropores with diameters of 1.4 and 1.7 nm, respectively (Fig. 1E). Compared to MIL-101-NH2, the BET surface areas and pore volumes of TC-MIL-101 and BC-MIL-101 significantly decrease while those of OC-MIL-101 only change slightly. It is reported that the porosity of MOF is reduced by the reaction with hydrochloric acid produced during the post synthesis [33].

|
Download:
|
| Fig. 1. Synthesis and structure characterizations of post-modified MIL-101 MOFs: (A) Schematic illustration for the post-synthesis of TC-MIL-101, BC-MIL-101 and OC-MIL-101 from MIL-101-NH2; (B) SEM images (scale bar = 100 nm); (C) XRD patterns; (D) 77 K N2 sorption isotherms; (E) Pore-size distribution by NLDFT method; (F) TGA curves. | |
{kind=link}
The thermal stability of the MOFs was examined by thermal gravimetric analysis (TGA, Fig. 1F). The weight loss before 200 ℃ of MIL-101-NH2 is due to the evaporation of guest molecules in pores and the weight loss between 200–290 ℃ is due to the leave of coordinated water and OH groups [31]. The significant weight loss between 290 ℃ and 320 ℃ can be attributed to the collapse of the crystal structure. The crystal decomposition temperatures of MIL-101-NH2, TC-MIL-101, BC-MIL-101 and OC-MIL-101 are 278, 324, 342 and 310 ℃ (Fig. S1 in Supporting information), respectively.
The chemical structures of the MIL-101 MOFs were characterized by fourier transform infrared spectroscopic (FT-IR) (Fig. 2A) and 1H NMR (Fig. 2B). In the FT-IR spectra, characteristic peaks of MIL-101-NH2 are observed at 1580−1660 cm−1 (N-H bending vibration), 1496 cm−1, 1433 cm−1 (stretching vibrations of O-C-O units) and 1257 cm-1 (stretching vibration of C-N) [28, 34]. Compared with MIL-101-NH2, the three modified MOFs show some peaks with frequency shift or intensity change in addition to the characteristic peaks of MIL-101-NH2. The intensity and frequency of peaks at 1580−1660 cm−1 are significantly changed because of the appearance of C=O stretching vibration in the newly formed amide groups. Moreover, the C-N stretching vibration of aromatic amines (1340 cm−1 and 1257 cm−1) become invisible or less prominent. Accordingly, a new characteristic N-H bending vibration was observed at 1303 cm−1 because of the formation of aromatic amides.

|
Download:
|
| Fig. 2. The compositional characterization of post-synthesized MIL-101 MOFs: (A) FT-IR spectra; (B) 1H NMR spectra of the digested products from various MIL-101 MOFs. | |
{kind=link}
The degree of post-synthetic modification was determined by 1H NMR after the digestion of MOFs in NaOH/D2O solution (Fig. 2B). The digested solution contained unmodified 2-aminoterephthalic acid and the corresponding amidoterephthalic acid. The chemical shifts of aromatic protons moved to downfield after the formation of aromatic amides. Thus, we can obtain the conversion rates by calculating and comparing the peak integration of the aromatic protons and substituent group protons. The conversion rates were determined as 65.8%, 63.1% and 53.5% for TC-MIL101, BC-MIL-101 and OC-MIL-101, respectively.
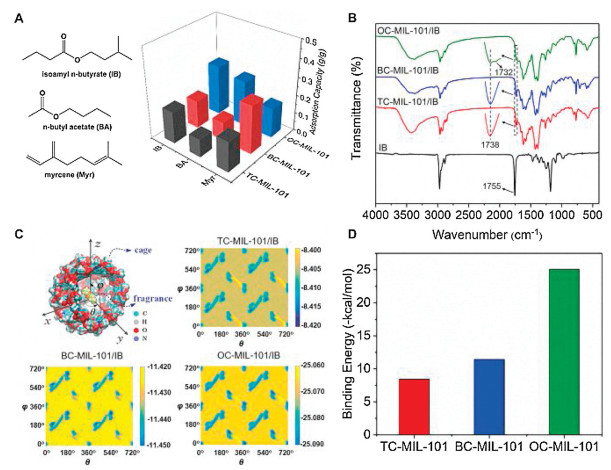
To examine the encapsulation capacities of modified MOFs, we selected several commonly used fragrances as model molecules, including myrcene (Myr), n-butyl acetate (BA) and isoamyl n-butyrate (IB). There is no apparent change in the structure and morphology of MOFs after loading fragrant molecules (Figs. S2 and S3 in Supporting information). The fragrance loading capacities were determined by comparing the TGA weight loss of the pristine MOFs and the corresponding fragrance-loaded MOFs (Fig. 3A and Table S1 in Supporting information). All the MOFs show high capability in encapsulation of fragrant molecules with a loading content of 0.10 g to 0.28 g fragrance per gram of MOFs (g/g, Table S1). Among them, the oxalic acid-functionalized OC-MIL-101 shows a 2−3-fold higher loading capacity of the two polar esters, IB and BA, than those of TC-MIL-101 and BC-MIL-101. This result is probably due to the relatively large surface area and the formation of strong interactions between polar molecules and polar pores of OC-MIL-101, which is consistent with our previous findings [9, 10]. The adsorption capacity of the terpenoid-type Myr on BC-MIL-101 is about 2-fold of those on other MOFs, which is probably affected by the π-π stacking interaction between the phenyl ring and unsaturated bonds.

|
Download:
|
| Fig. 3. Encapsulation of volatile molecules in post-synthesized MIL-101 MOFs and the host-guest interaction analysis: (A) The chemical structures of representative guest molecules, isoamyl n-butyrate (IB), n-butyl acetate (BA) and myrcene (Myr), and their encapsulation capacity in different MOFs; (B) The FT-IR of IB and IB-loaded MOFs; (C) The schematic of an isolated MIL-101 cage capturing an IB molecule in its center and the direction dependence of binding energies of TC-MIL-101 with the IB molecule; (D) The binding energies of IB to different MOFs. | |
{kind=link}
To study the molecular interactions between MOFs and fragrances, the FT-IR spectra of MOFs/fragrances and free fragrances were recorded (Fig. 3B, Figs. S4 and S5 in Supporting information). The FT-IR spectra of IB and IB-loaded MOF are shown in Fig. 3B as representatives. Compared to pristine MOF (Fig. 2A), all the IB-loaded MOFs (MOF/IB) show a new peak at around 1738 cm−1, which can be attributed to the ester carbonyl C=O stretching of IB molecule. Compared to free IB, the C=O stretching peak in MOF/IB shows a redshift by 17 cm−1, suggesting the formation of hydrogen bonds in the C=O-H+ moiety between amide/amine and ester group [9, 10]. OC-MIL-101 shows another C=O stretching peak at about 1732 cm−1, which is probably due to the formation of strong hydrogen bonds between carboxyl and ester group. These phenomena were also observed in BA-loaded MOF, further confirming the existence of hydrogen bond interaction between polar fragrance and MOFs (Fig. S4). The loading of the terpenoid-type Myr in MOFs shows redshift of the C=C stretching frequency at 1595 cm−1, probably due to π-π stacking interaction [35]. As a result, the intensity of peaks at 1615 cm−1 of MOFs/Myr are obviously strengthen compared to those of MOFs (Fig. S5).
The host-guest interactions of fragrance IB with the cage-like moieties of TC-MIL-101, BC-MIL-101 and OC-MIL-101 were further studied by molecular dynamics calculation using Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) [36]. Isolated MIL-101 cages was built by Materials Studio 2019. As Fig. 3C shows, IB binds to a simulated isolated cage of MIL-101 and locates in its geometry center. The binding energies of IB and the cage were mapped by rotating in two orthogonal directions both from 0 to 360 degrees and replicated to 720 degrees for visual consideration. IB exhibits different direction dependence of binding energies with TC-MIL-101, BC-MIL-101 and OC-MIL-101. The IB molecule is perpendicular to the normal of the pentagon window when the binding energies reach the minimum for all the three cage-like MIL-101 molecules. In this way, the stable locking of IB can be achieved geometrically. The average binding energy (Eb) of IB to different MOFs was determined as -25.068 kcal/mol for OC-MIL-101/IB, -8.405 kcal/mol for TC-MIL-101/IB, and -11.428 kcal/mol for BC-MIL-101/IB (Fig. 3D). The results show that OC-MIL-101 has the most potent host-guest interactions with IB. It is in agreement with the observation of FT-IR that the formation of hydrogen bonds reduces the binding energy [9, 37]. Interestingly, the IB encapsulation capacities in different MOFs are also correlated with their average binding energies (Fig. 3A and Table S1). All these results suggest that the encapsulation capability of guest molecules in porous MOF is affected by the host-guest interactions, and the strong interaction may enhance the loading capacity.
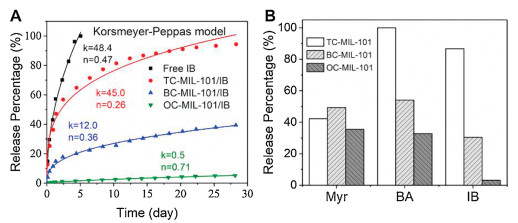
We propose that the host-guest interactions between the guest molecules and MOF may also regulate the release profiles. To test this assumption, we studied the release profiles of Myr, BA or IB loaded MOFs to compare with free fragrances. We found that free fragrances were completed released in a couple of days, while all the fragrances loaded in MOFs showed sustained release profiles (Fig. 4A, Figs. S6 and S7 in Supporting information). The releasing amount of TC-MIL-101/IB over two weeks is around 3.06 and 27.4 times that of BC-MIL-101 and OC-MIL-101, respectively. The stronger host-guest interactions lead to a more sustained release of guest molecules. This tendency on release profiles is also observed on BA-loaded MOFs (Fig. S6). There is no apparent difference in the release profiles of terpene-type Myr among all the MOFs which may due to the small interaction differences between the nonpolar molecule and MOFs (Fig. S7).

|
Download:
|
| Fig. 4. Sustained release of volatile molecules from post-synthesized MIL-101-type MOFs: (A) Release profiles and kinetics of IB and IB-loaded MOFs fitted by Korsmeyer-Peppas mathematical model at 25 ℃; (B) Release percentage of different volatile molecules from MOFs over two weeks at 25 ℃. | |
{kind=link}
We next studied the release kinetics of MOFs/fragrances using four mathematic models, including zero-order (yt/y∞ = kt + c), first-order (yt/y∞ = 1 - exp[-kt]), Higuchi (yt/y∞ = kt0.5), and Korsmeyer-Peppas (yt/y∞ = ktn + c) models. The release curves of MOFs/IB can be well fitted by the Korsmeyer-Peppas model (Fig. 4B and Fig. S8 in Supporting information) [38]. The release rate constant k was determined as 48.4 for free IB, 45.0 for TC-MIL-101/IB, 12.0 for BC-MIL-101/IB, and 0.5 for OC-MIL-101/IB. Accordingly, the half release time (t50) was calculated as 1.1 days for free IB, 2.1 days for TC-MIL-101/IB, 55.5 days for BC-MIL-101/IB, and 674.8 days for OC-MIL-101/IB (Table S2 in Supporting information). This result suggests that the release of volatile molecules can be adjusted from days to months and even over a year by controlling over the host-guest interactions. The parameter n of the Korsmeyer-Peppas equation can be used to determine the diffusion types. For n < 0.45, it is a diffusion-controlled release; for 0.45 < n < 0.89, it represents synergistic transport; and for n > 0.89, it follows swelling controlled release. The index n for TC-MIL-101/IB and BC-MIL-101/IB are 0.26 and 0.36, respectively, indicating that the release mechanisms of TC-MIL-101/IB and BC-MIL-101/IB are diffusion-controlled release. The index n for OC-MIL-101 is 0.71, showing a synergistic transport release mechanism. The release curves of BA and Myr were also fitted by the Korsmeyer-Peppas model (Figs. S9 and S10 in Supporting informatoin). All the release mechanisms of MOFs/BA are synergistic transport, while the release mechanisms of Myr from MOFs/Myr are diffusion release. These results suggested that the diffusion mechanism of volatile molecules from MOF is affected by both the guest molecules and the host carrier.
In conclusion, we synthesized three pore-functionalized MIL-101-type MOFs by postsynthetic modification. The functionalized MOFs have different guest-host interactions with volatile guest molecules and exhibit various encapsulation capacities and sustained release profiles. The release profiles are correlated with the binding energies between the guest volatiles and pores in MOFs. Chemical engineering the pore surface of MOF is capable of tuning the release of encapsulated molecules from days to several months and even over a year. Thus, we believe that the post-modification of MOF holds excellent potential for application in encapsulation and sustained release of volatile molecules.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work is supported by the National Key Research and Development Program of China (No. 2016YFA0200301), the National Natural Science Foundation of China (Nos. 21875211 and 51603181). The authors thank the High-performance Computing Platform of Peking University for the support of simulation. The authors also thank Mrs. Na Zheng in the State Key Laboratory of Chemical Engineering at Zhejiang University for help on SEM experiments.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.035.
| [1] |
B. Adorjan, G. Buchbauer, Flavour Frag. J. 25 (2010) 407-426. DOI:10.1002/ffj.2024 |
| [2] |
A. Haehner, H. Maass, I. Croy, T. Hummel, Flavour Frag. J. 32 (2017) 24-28. DOI:10.1002/ffj.3339 |
| [3] |
C. Chrea, D. Grandjean, S. Delplanque, et al., Chem. Senses 34 (2009) 49-62. |
| [4] |
A. Herrmann, Angew. Chem. Int. Ed. 46 (2007) 5836-5863. DOI:10.1002/anie.200700264 |
| [5] |
B. Pena, C. Panisello, G. Areste, R. Garcia-Valls, T. Gumi, Chem. Eng. J. 179 (2012) 394-403. DOI:10.1016/j.cej.2011.10.090 |
| [6] |
Y. Liu, K. Liu, M. Zhao, et al., React. Funct. Polym. 132 (2018) 138-144. DOI:10.1016/j.reactfunctpolym.2018.09.021 |
| [7] |
R. Tekin, N. Bac, J. Warzywoda, A. Sacco, Microporous Mesoporous Mater. 215 (2015) 51-57. DOI:10.1016/j.micromeso.2015.05.020 |
| [8] |
B. Strzemiecka, M. Kasperkowiak, M. Lozynski, D. Paukszta, A. Voelkel, Microporous Mesoporous Mater. 161 (2012) 106-114. DOI:10.1016/j.micromeso.2012.05.024 |
| [9] |
Y. Liu, Y. Wang, J. Huang, et al., AlChE J. 65 (2019) 491-499. DOI:10.1002/aic.16461 |
| [10] |
B. Zhang, J. Huang, K. Liu, et al., Ind. Eng. Chem. Res. 58 (2019) 19767-19777. DOI:10.1021/acs.iecr.9b04214 |
| [11] |
A.M. Shultz, O.K. Farha, J.T. Hupp, S.T. Nguyen, J. Am. Chem. Soc. 131 (2009) 4204-4205. DOI:10.1021/ja900203f |
| [12] |
A. Schneemann, V. Bon, I. Schwedler, et al., Chem. Soc. Rev. 43 (2014) 6062-6096. DOI:10.1039/C4CS00101J |
| [13] |
Z. Han, W. Shi, P. Cheng, Chin. Chem. Lett. 29 (2018) 819-822. DOI:10.1016/j.cclet.2017.09.050 |
| [14] |
X. Wang, W. Chen, H. Qi, et al., Chem. Eur. J. 23 (2017) 7990-7996. DOI:10.1002/chem.201700474 |
| [15] |
Z. Hu, B.J. Deibert, J. Li, Chem. Soc. Rev. 43 (2014) 5815-5840. DOI:10.1039/C4CS00010B |
| [16] |
K. Sheng, H. Lu, A. Sun, et al., Chin. Chem. Lett. 30 (2019) 895-898. DOI:10.1016/j.cclet.2019.01.027 |
| [17] |
D. Zhao, S. Tan, D. Yuan, et al., Adv. Mater. 23 (2011) 90-93. DOI:10.1002/adma.201003012 |
| [18] |
Z. Dong, Y. Sun, J. Chu, X. Zhang, H. Deng, J. Am. Chem. Soc. 139 (2017) 14209-14216. DOI:10.1021/jacs.7b07392 |
| [19] |
J. Della Rocca, D. Liu, W. Lin, Acc. Chem. Res. 44 (2011) 957-968. DOI:10.1021/ar200028a |
| [20] |
X. Cui, K. Chen, H. Xing, et al., Science 353 (2016) 141-144. DOI:10.1126/science.aaf2458 |
| [21] |
X. Kong, H. Deng, F. Yan, et al., Science 341 (2013) 882-885. DOI:10.1126/science.1238339 |
| [22] |
M. Ding, X. Cai, H.L. Jiang, Chem. Sci. 10 (2019) 10209-10230. DOI:10.1039/C9SC03916C |
| [23] |
S. Yang, L. Peng, D. Sun, et al., Chem. Sci. 10 (2019) 4542-4549. DOI:10.1039/C9SC00135B |
| [24] |
H. Jiang, D. Feng, T. Liu, J. Li, H. Zhou, J. Am. Chem.Soc. 134 (2012) 14690-14693. DOI:10.1021/ja3063919 |
| [25] |
N.C. Burtch, H. Jasuja, K.S. Walton, Chem. Rev. 114 (2014) 10575-10612. DOI:10.1021/cr5002589 |
| [26] |
M. Kim, J.F. Cahill, H. Fei, K.A. Prather, S.M. Cohen, J. Am. Chem. Soc. 134 (2012) 18082-18088. DOI:10.1021/ja3079219 |
| [27] |
K.K. Tanabe, S.M. Cohen, Chem. Soc. Rev. 40 (2011) 498-519. DOI:10.1039/C0CS00031K |
| [28] |
F. Zhou, J. Zhou, X. Gao, C. Kong, L. Chen, RSC Adv. 7 (2017) 3713-3719. DOI:10.1039/C6RA25396B |
| [29] |
Z. Wang, K.K. Tanabe, S.M. Cohen, Chem. Eur. J. 16 (2010) 212-217. DOI:10.1002/chem.200902158 |
| [30] |
J. Wang, X. Huang, H. Gao, A. Li, C. Wang, Chem. Eng. J. 350 (2018) 164-172. DOI:10.1016/j.cej.2018.05.190 |
| [31] |
Y. Lin, C. Kong, L. Chen, RSC Adv. 2 (2012) 6417-6419. DOI:10.1039/c2ra20641b |
| [32] |
H. Liu, D. Ramella, P. Yu, Y. Luan, RSC Adv. 7 (2017) 22353-22359. DOI:10.1039/C7RA00296C |
| [33] |
H. Hintz, S. Wuttke, Chem. Mater. 26 (2014) 6722-6728. DOI:10.1021/cm502920f |
| [34] |
Q. Xie, Y. Li, Z. Lv, et al., Sci. Rep. 7 (2017) 3316. DOI:10.1038/s41598-017-03526-x |
| [35] |
P. Karthik, R. Vinoth, P. Zhang, et al., ACS Appl. Energy Mater. 1 (2018) 1913-1923. DOI:10.1021/acsaem.7b00245 |
| [36] |
S. Plimpton, J. Comput. Phys. 117 (1995) 1-19. DOI:10.1006/jcph.1995.1039 |
| [37] |
J. Huang, B. Wu, Z. Zhou, et al., Nanomedicine 21 (2019) 102058. DOI:10.1016/j.nano.2019.102058 |
| [38] |
R.W. Korsmeyer, R. Gurny, E. Doelker, P. Buri, N.A. Peppas, Int. J. Pharm. 15 (1983) 25-35. DOI:10.1016/0378-5173(83)90064-9 |