 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory of Medicinal Chemical Biology & Tianjin Key Laboratory of Protein Sciences, College of Life Sciences, Nankai University, Tianjin 300071, China
Cancer immunotherapy is attracting increasing attention as it avoids the shortcomings of traditional methods of cancer treatment [1]. Therapeutic cancer vaccine is an important type of cancer immunotherapy [2]. In the development of cancer vaccines, Mucin 1 (MUC1), a transmembrane glycoprotein which is abnormally glycosylated and overexpressed in various epithelial cell carcinoma, has been proven to be a promising target [3]. The extracellular domain of MUC1 consists of a variable number of tandem repeats, which are a sequence of 20 amino acids (i.e., HGVTSAPDTRPAPGSTAPPA). Five Ser/Thr residues of this sequence may be O-glycosylated [4]. Tumor cells are characterized by the overexpression of MUC1 and truncated and over-sialylated glycosylation pattern [5, 6]. The common types of glycan side chains expressed on MUC1 are Tn antigen, T antigen, and their sialylated forms [7]. However, MUC1 glycopeptide-based antigens are T-cell independent and tolerated by immune systems; Thus, they are weakly immunogenic [8-10]. To solve these problems, many studies have been conducted to develop effective MUC1-based cancer vaccines [11-20]. Nevertheless, the construction of vaccines with highly effective immune response is still a challenge.
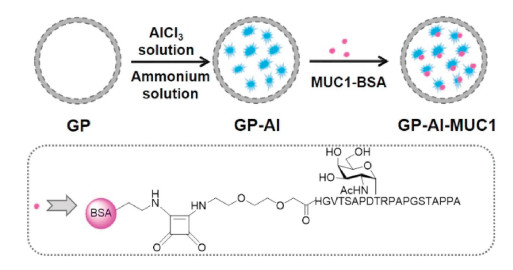
GP-Al are hybrid nanoparticles with the size of 2–4 μm, which were prepared by sealing the alum colloid inside the β-glucan particles (GPs). In our previous study, we have prepared GP-Al hybrid particles for the first time and demonstrated these particles can be applied for loading proteins by simple mixing that avoids manipulation of the proteins with any complex chemical or physical operations. The prepared GP-Al particles, which specifically targeted to antigen-presenting cells (APCs), can intensively activate DC maturation to secret various cytokines. The very uniform size, strong immune stimulation functions of GP-Al particles indicate their potential application as immune activator for cancer immunotherapy [21].
Herein, novel MUC1 glycopeptide-based vaccine was constructed with GP-Al hybrid particles as the immunoenhancer. As GP-Al can only load macromolecular proteins, not small molecules [21]. In order to prepare the vaccine, small molecule antigen MUC1 glycopeptide was first covalently linked with carrier protein BSA here. Then they were absorbed by GP-Al particles through electrostatic adsorption forming the potential vaccine (named as GP-Al-MUC1). Immunological results showed the constructed vaccine induced high level of IgG antibodies and cytokines. Moreover, it enhanced the ability of cytotoxic T lymphocytes (CTL) to effectively kill tumor cells. All of the above results indicated GP-Al particles were promising carrier and adjuvant for MUC1 antigen based tumor vaccines.
To prepare GP-Al hybrid particles, the β-glucan particles (GPs) were firstly prepared from yeast by removing the protein and DNA through pre-processing with NaOH. Then the prepared GPs were steeped in AlCl3 solution (0.125 mol/L) followed by centrifugation to remove the excess aluminum solution outside the GPs. The aluminum inside the GPs was transformed into colloid with excess ammonium solution. To increase the superficial area of the aluminum colloid inside the particles, the particles were sonicated for half an hour to disperse the colloid into nanometer size to form GP-Al particles. The GP-Al-MUC1 was prepared by a simple mixing of GP-Al and MUC1-BSA (Fig. 1).

|
Download:
|
| Fig. 1. Schematic demonstration of the preparation of MUC1 vaccine loaded on GP-Al particles. | |
{kind=link}
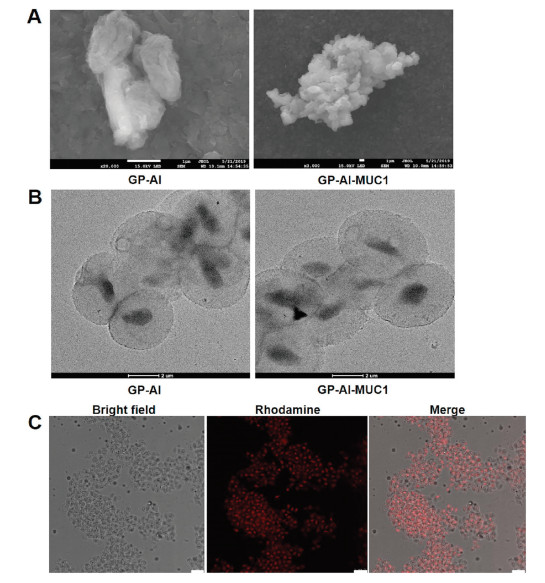
The prepared GP-Al and GP-Al-MUC1 particles were characterized. The morphology of the prepared GP-Al and GP-Al-MUC1 particles was first observed through SEM. As shown in Fig. 2A, both particles exhibited a wrinkled surface during the dehydration process. TEM images showed that the particles were well dispersed and had very uniform particle size, approximately 2–4 μm (Fig. 2B). The dispersed distribution of the GP-Al particles and the antigen-loaded GP-Al-MUC1 particles was also demonstrated via confocal fluorescence microscopy (Fig. 2C), the results showed the particle size was very uniform which was indispensable for the uptake by the APCs. Meanwhile, the aluminum contents in GP-Al and GP-Al-MUC1 particles determined by ICP-OES were both 1.0% ± 0.1%.

|
Download:
|
| Fig. 2. Characterization of the GP-Al and GP-Al-MUC1 particles. (A) SEM images of the GP-Al and GP-Al-MUC1 particles. Scale bar: 1 μm. (B) TEM images of the GP-Al and GP-Al-MUC1 particles. Scale bar: 2 μm. (C) Confocal images of the GP-Al-MUC1 particles with rhodamine-labeled MUC1-BSA encapsulated inside GP-Al. Scale bar: 10 μm. | |
{kind=link}
Antigen loading efficiency was measured. The GP-Al particles were mixed with MUC1-BSA together, then the loading capacity was calculated by measuring the unloaded MUC1-BSA in the supernatant by BCA method. The loading capacity of approximately 4% (4 mg MUC1-BSA protein/100 mg GP-Al-MUC1) was received according to the procedure. The antigen release rate of GP-Al-MUC1 particles was performed by monitoring the release of MUC1-BSA from the particles in a shaker at 37℃. The results showed only 18% and 32% of the entrapped MUC1-BSA was released from the particles over 24 h and 48 h respectively, and 55% of the entrapped MUC1-BSA was released over 72 h. MUC1-BSA release was sustained for 4 days, which indicated the particles had good antigen retention ability (Fig. S1 in Supporting information).
Immunological evaluation of the vaccine candidates was then conducted. Female BALB/c mice were immunized every two weeks for four times via subcutaneous injection. Sera were collected one week after the last immunization. The antibody level was evaluated with MUC1 antigen-specific ELISA. As shown in Fig. 3, GP-Al-MUC1 and MUC1-BSA groups remarkably increased anti-MUC1 IgG antibody titers, while GP-Al and PBS groups almost could not elicit detectable anti-MUC1 antibodies. Compared with MUC1-BSA group, GP-Al-MUC1 elicited a higher IgG titer which indicated GP-Al may be served as the adjuvant to enhance the immunogenicity of MUC1 antigen.

|
Download:
|
| Fig. 3. MUC1-specific IgG antibody level of the mice immunized by GP-Al, MUC1-BSA and GP-Al-MUC1 analyzed using ELISA, PBS was used as control. MUC1 glycopeptide was coated on the ELISA plates. Data are shown as mean±SD. | |
{kind=link}
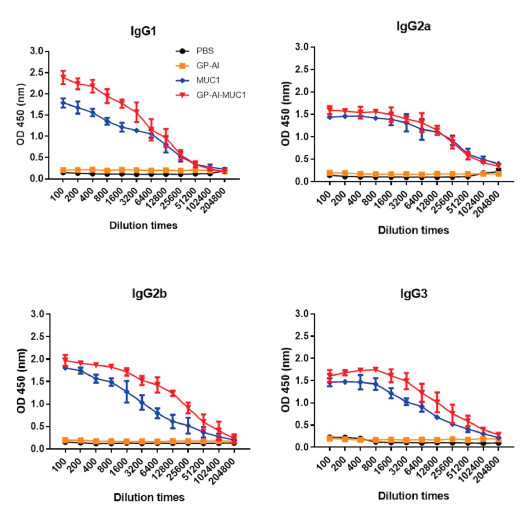
The antibody isotypes of IgG1, IgG2 a, IgG2 b and IgG3 were then evaluated. As shown in Fig. 4, GP-Al-MUC1 distinctly enhanced the production of IgG1, IgG2 a, IgG2 b and IgG3 than MUC1-BSA, this is in agreement with previous IgG results. Moreover, GP-Al-MUC1 mainly induced the IgG1 rather than IgG2 a antibody. As IgG1 level indicated Th2-biased immune response, and IgG2 a level meant Th1-biased immune response [22, 23]. This result showed GP-Al-MUC1 mainly induced Th2-type immune responses.

|
Download:
|
| Fig. 4. Antibody isotypes (IgG1, IgG2 a, IgG2 b and IgG3) analysis of the antisera induced by the vaccine candidates through ELISA. Data are shown as mean±SD. | |
{kind=link}
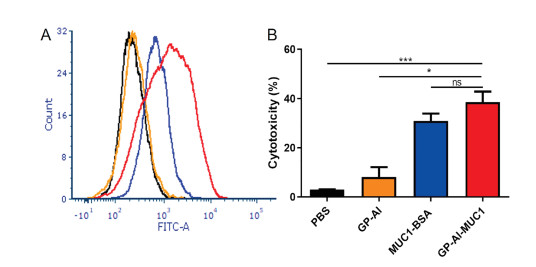
The binding affinity of the antisera induced by the vaccines with tumor cells was further analyzed [24, 25]. MUC1-expressing MCF-7 tumor cells was applied, and the result was evaluated by flow cytometry (FACS). Briefly, antisera from immunized mice were incubated with MCF-7 cells and cultured with FITC-labeled rabbit anti-mouse antibody. As shown in Fig. 5A, among all the groups, the sera from GP-Al-MUC1 showed the highest binding with MCF-7 cells.

|
Download:
|
| Fig. 5. (A) Binding of the antisera elicited by GP-Al (orange), MUC1-BSA (blue) and GP-Al-MUC1 (red) with MCF-7 cells analyzed by FACS. The sera collected from the PBS group (black) were used as control. (B) CDC effect of the antisera induced by GP-Al, MUC1-BSA and GP-Al-MUC1 to MCF-7 cells. | |
{kind=link}
To further assess the function of the antibodies induced by the vaccine candidates, CDC effect of the antisera was analyzed. MUC1-expressing MCF-7 cells were incubated with the antisera, and rabbit complement was then added. The cytotoxicity of tumor cells was assessed by CCK-8 method. As shown in Fig. 5B, the antisera induced by GP-Al-MUC1 mediated the most efficient CDC to MCF-7 cells, which was in accordance with previous antibody results. This further demonstrated that GP-Al was crucial for the production of effect antibodies.
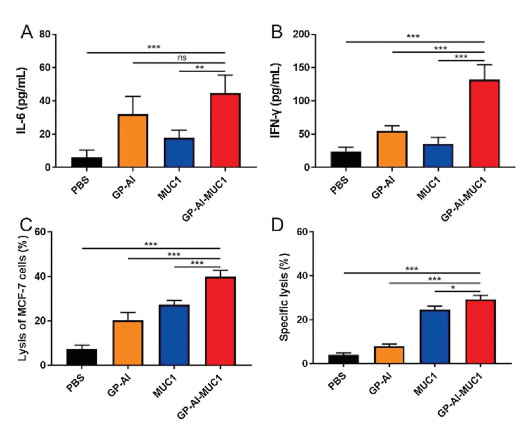
In order to further investigate the immune effects induced by the vaccine candidates, the cytokines of the sera from immunized mice were analyzed. IL-6, which can promote the maturation and differentiation of B cells to secret antibodies, is a Th2-type cytokine; IFN-γ, which can enhance the cytotoxicity of cytotoxic T lymphocytes (CTL), are Th1-type cytokines [26, 27]. As shown in Figs. 6A and B, IL-6 and IFN-γ obviously increased in the mice immunized with GP-Al-MUC1 compared with those injected with MUC1-BSA. These results meant that immunization of the vaccines contained GP-Al could increase the expression of Th1 and Th2 type cytokines.

|
Download:
|
| Fig. 6. The secretion of (A) IL-6 and (B) IFN-γ in the sera was analyzed by ELISA. (C) CTL-mediated cytotoxicity in vitro was analyzed via LDH cytotoxicity assay. (D) CTL killing assays in vivo was performed by intravenous injection of CFSE-labelling splenocytes into immunized mice and detected by FACS. Data are shown as mean±SD. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001. | |
{kind=link}
Cytotoxic T lymphocytes (CTL) can kill cancer cells to induce tumor regression [28]. To analyze in vitro CTL effect, splenocytes were collected from the mice immunized with the vaccine candidates. CD8+ T cells were isolated to co-incubate with MCF-7 cells in the ratio of 50:1. The cytotoxicity was analyzed by LDH Cytotoxicity Assay Kit. As shown in Fig. 6C, MUC1-BSA induced stronger cytotoxicity than GP-Al, while GP-Al-MUC1 elicited more powerful cytotoxicity compared with MUC1-BSA. These results showed GP-Al-MUC1 could induce decent CTL effect to kill MCF-7 cells, which indicated it had the potential to induce protective and therapeutic effects against cancer.
Antigen-specific in vivo CTL responses were then performed. Splenocytes collected from normal mice were divided into two parts. One part was stimulated with MUC1 glycopeptide, while another part with nothing. The MUC1 glycopeptide pulsed splenocytes were labeled with CFSE (5-carboxyfluorescein diacetate succinimidyl ester) at 10 μmol/L (CFSEhigh), and nothing pulsed splenocytes with CFSE at 1 μmol/L (CFSElow). Then the same number of CFSEhigh cells and CFSElow cells were intravenously injected into immunized mice. The ratio of CFSE-labelled cells in spleen was analyzed by FACS. As shown in Fig. 6D, the strongest response was observed against the MUC1 glycopeptide in GP-AL-MUC1 group, which was consistent with previous in vitro CTL results.
Generally speaking, GP-Al was demonstrated to be used as carrier system and adjuvant for the construction MUC1 based antitumor vaccine. Firstly, GP-Al can load MUC1 antigen protein conjugate just by a very simple mixing procedure, which avoids any cumbersome operations. Compared with the immunization procedures of other adjuvants, such as Freund's adjuvant which needs a complex emulsification when used [29], GP-Al is very easy to operate. Moreover, the result of in vitro release experiment indicates the antigen protein can be trapped inside GP-Al particles for more than three days, the long-lasting stimulation time may contribute to their strong immune responses. Secondly, GP-Al can enhance the production of antibodies and induce CTL effect, which means the excellent adjuvant effect of GP-Al. We can find clues to GP-Al adjuvant activity from previous report. The β-1, 3-d-glucan structure on the surface of GPs can serve as a PAMP to activate dectin-1 receptor on DCs, which stimulate innate and adaptive immune responses [28, 30, 31]. Compared with our previous work where GP-Al can only load macromolecular proteins [21], we here demonstrate GP-Al can be used for loading small molecule fragments. It should be noted that the small molecule fragments must conjugate with protein such as BSA when they are loaded on GP-Al which needs further optimization. Overall, this work suggests that GP-Al can be used as an efficient vaccine carrier system and adjuvant for the development of small molecule antigens based cancer vaccines.
In this study, small molecule MUC1 antigen based antitumor vaccine was constructed by loading its conjugate with the protein of BSA on GP-Al. In the prepared vaccine, GP-Al particles served as antigen delivery system and vaccine adjuvant. Immunological evaluation revealed that the designed vaccine could elicit high levels of antibodies and cytokines. Antisera induced by the vaccine can strongly bind with tumor cells and induced CDC effect. Meanwhile, the vaccine can enhance the cytotoxicity of CTL to kill tumor cells. In general, the study provided a novel strategy to construct small molecule antigens based cancer vaccines using GP-Al nanoparticles for the development of cancer immunotherapy.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 22077068), the National Key R & D Program of China (No. 2018YFA0507204), the NCC Fund (No. NCC2020FH12), the Natural Science Foundation of Tianjin (No. 19JCQNJC05300), the Fundamental Research Funds for the Central Universities. All animal experiments were approved by the Animal Ethical and Welfare Committee of Nankai University and conducted in accordance with Nankai University Experimental Animal Ethics Committee.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.035.
| [1] |
S.A. Rosenberg, J.C. Yang, N.P. Restifo, Nat. Med. 10 (2004) 909-915. DOI:10.1038/nm1100 |
| [2] |
I. Melero, G. Gaudernack, W. Gerritsen, et al., Nat. Rev. Clin. Oncol. 11 (2014) 509-524. DOI:10.1038/nrclinonc.2014.111 |
| [3] |
R.M. Wilson, S.J. Danishefsky, J. Am. Chem. Soc. 135 (2013) 14462-14472. DOI:10.1021/ja405932r |
| [4] |
W.H. Li, Y.M. Li, Chem. Rev. 120 (2020) 11420-11478. DOI:10.1021/acs.chemrev.9b00833 |
| [5] |
J.A. Schroeder, A.A. Masri, M.C. Adriance, et al., Oncogene 23 (2004) 5739-5747. DOI:10.1038/sj.onc.1207713 |
| [6] |
J. Zhao, G. Hu, Y. Huang, et al., Chin. Chem. Lett. 32 (2021) 1331-1340. DOI:10.1016/j.cclet.2020.10.013 |
| [7] |
N. Gaidzik, U. Westerlind, H. Kunz, Chem. Soc. Rev. 42 (2013) 4421-4442. DOI:10.1039/c3cs35470a |
| [8] |
S.J. Danishefsky, J.R. Allen, Angew. Chem. Int. Ed. 39 (2000) 836-863. DOI:10.1002/(SICI)1521-3773(20000303)39:5<836::AID-ANIE836>3.0.CO;2-I |
| [9] |
D. Feng, A.S. Shaikh, F. Wang, ACS Chem. Biol. 11 (2016) 850-863. DOI:10.1021/acschembio.6b00084 |
| [10] |
S.J. Danishefsky, J.R. Allen, Angew. Chem. Int. Ed. 39 (2000) 836-863. DOI:10.1002/(SICI)1521-3773(20000303)39:5<836::AID-ANIE836>3.0.CO;2-I |
| [11] |
Y. Gao, Z.Y. Sun, Z.H. Huang, et al., Chem. Eur. J. 20 (2014) 13541-13546. DOI:10.1002/chem.201404013 |
| [12] |
Y. Shao, Z.Y. Sun, Y. Wang, et al., ACS Appl. Mater. Interfaces 10 (2018) 9310-9314. DOI:10.1021/acsami.8b00312 |
| [13] |
Z. Yin, H.G. Nguyen, S. Chowdhury, et al., Bioconjugate Chem. 23 (2012) 1694-1703. DOI:10.1021/bc300244a |
| [14] |
I. Companon, A. Guerreiro, V. Mangini, et al., J. Am. Chem. Soc. 141 (2019) 4063-4072. DOI:10.1021/jacs.8b13503 |
| [15] |
J.J. Wu, W.H. Li, P.G. Chen, et al., Chem. Commun. 54 (2018) 9655-9658. DOI:10.1039/C8CC04860F |
| [16] |
X.G. Yin, X.Z. Chen, W.M. Sun, et al., Org. Lett. 19 (2017) 456-459. DOI:10.1021/acs.orglett.6b03591 |
| [17] |
M. Glaffig, N. Stergiou, E. Schmitt, et al., ChemMedChem 12 (2017) 722-727. DOI:10.1002/cmdc.201700254 |
| [18] |
F. Broecker, S. Gotze, J. Hudon, et al., J. Med. Chem. 61 (2018) 4918-4927. DOI:10.1021/acs.jmedchem.8b00312 |
| [19] |
X. Wu, Z. Yin, C. McKay, et al., J. Am. Chem. Soc. 140 (2018) 16596-16609. DOI:10.1021/jacs.8b08473 |
| [20] |
Z. Zhou, H. Lin, C. Li, et al., Chin. Chem. Lett. 29 (2018) 19-26. DOI:10.1016/j.cclet.2017.09.047 |
| [21] |
H. Liu, Z. Jia, C. Yang, et al., Biomaterials 167 (2018) 32-43. DOI:10.1016/j.biomaterials.2018.03.014 |
| [22] |
M. Skwarczynski, M. Zaman, C.N. Urbani, et al., Angew. Chem. Int. Ed. 49 (2010) 5742-5745. DOI:10.1002/anie.201002221 |
| [23] |
S.S. Seregin, D.M. Appledorn, A.J. McBride, et al., Mol. Ther. 17 (2009) 685-696. DOI:10.1038/mt.2008.297 |
| [24] |
S. Ingale, M.A. Wolfert, J. Gaekwad, et al., Nat. Chem. Biol. 3 (2007) 663-667. DOI:10.1038/nchembio.2007.25 |
| [25] |
Z. Yin, X. Wu, K. Kaczanowska, et al., ACS Chem. Biol. 13 (2018) 1668-1676. DOI:10.1021/acschembio.8b00313 |
| [26] |
T. Osada, M.A. Morse, H.K. Lyerly, et al., Int. Immunol. 17 (2005) 1143-1155. DOI:10.1093/intimm/dxh292 |
| [27] |
K.L. Knutson, M.L. Disis, Cancer Immunol. Immunother. 54 (2005) 721-728. DOI:10.1007/s00262-004-0653-2 |
| [28] |
Z. Yang, M. Xu, Z. Jia, et al., Biomaterials 134 (2017) 51-63. DOI:10.1016/j.biomaterials.2017.04.035 |
| [29] |
H.Y. Qin, M.W. Sadelain, C. Hitchon, et al., J. Immunol. 150 (1993) 2072-2080. |
| [30] |
H. Huang, G.R. Ostroff, C.K. Lee, et al., Infect. Immun. 77 (2009) 1774-1781. DOI:10.1128/IAI.00086-09 |
| [31] |
S. Leibundgut-Landmann, F. Osorio, G.D. Brown, et al., Blood 112 (2008) 4971-4980. |