 2021, Vol. 32
2021, Vol. 32 


b CAS Center for Excellence in Nanoscience, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology (NCNST), University of Chinese Academy of Sciences, Beijing 100190, China
The emergence of molecular fluorescent light-up probes that can specifically turn on their fluorescent in the presence of targets has open up a new opportunity to advance biosensing and bioimaging. As compared to the conventional fluorescent molecular probes, the ones with fluorescence light-up characteristics are more intelligent and hold the advantages of less false positive signals and larger target-to-background ratios.
Aggregation-induced emission (AIE) is a unique photophysical phenomenon, which effectively overcomes the aggregation-caused quenching (ACQ) problem of traditional fluorescent dyes [1, 2]. AIEgen have recently attracted great research interest in the areas of cell imaging, biosensing and diagnosis due to the excellent performance of strong photobleaching resistance, high signal-to-noise ratio and low cytotoxicity. Propeller-shaped fluorogens, such as tetraphenylethene (TPE) and silole derivatives [3], have almost no fluorescence emission in the molecularly dissolved state but induced to emit strong fluorescence in the aggregation state owing to the restriction of intramolecular rotation (RIR) and prohibition of energy dissipation through nonradiative channels. At present, the aggregation-driven florescence mechanism has been exploited to develop AIE-based bioresponsive FL probes [4, 5]. Many attempts have been made to tailor the emission properties of TPE via substitution or functionalization and to expand their applications in optical devices, sensors and biological imaging.
Inspired by nature, peptides have been widely used as a regulator in fabrication of biomimetic functional materials [6, 7], Peptide self-assembly is a promising approach for constructing and modulating supramolecular structures [7-9]. The assembled peptides have shown potential in bioimaging [10] and diagnosis [11] due to its various fabricated nanostructures resulting in multifunction capability and good biocompatibility. Recently, our group has reported an assembly/aggregation-induced retention (AIR) effect [12-14] in the targeting region in vivo, which can amplify the sensitivity and specificity for bioimaging [15, 16]. In addition, the assembled nanostructures can be modulated based on multiple weak bond interactions in order to obtain various biofunctions [17, 18]. Peptide-induced assembly of AIE molecules will not only help insight into tunable emission of AIE species, but also direct design of a new type of nanoarchitectonics and materials with endowed biofunctions, which has presented potential for applications in biomedicine and optoelectronic devices [19-21].
Herein, we exploit a bioactivated in vivo assembly (BIVA) peptide-tetraphenylethylene (TPE) probe with controllable assembled nanostructures for enhanced imaging of pancreatic tumor cell. Remarkably, by regulating the assembly of TPE through self-assembly peptide sequence, the intramolecular movement of TPE can be strongly restricted, so that the probe has a higher fluorescence signal output. On the other hand, the peptide can induce TPE to form various nanostructures for enhancing retention and accumulation on the surface of cells with better imaging window. We envision that the peptide-induced AIEgen self-assembly offers a new strategy to develop highly sensitive and efficient fluorescent light-up probes with designed nanostructures.
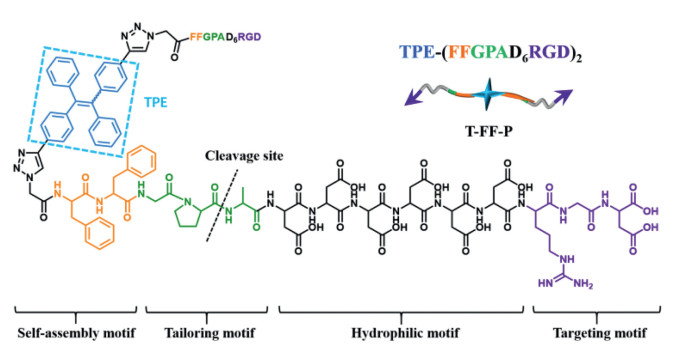
TPE, as a classic AIE fluorogen, emits strongly in aggregate state but shows very weak fluorescence in dilute solutions [22]. This is due to the propeller-shaped structure of TPE, and the dynamic rotations of the phenyl rings nonradiatively deactivated their excited states in solution. In the aggregate state, the RIR due to the physical constraint in the aggregates opens the radiative decay channel [23]. As shown in Fig. 1, the peptide-TPE probe (T-FF-P): TPE-(FFGPAD6RGD)2 with four moieties was developed. The modular design of the molecule including (i) a targeting motif: Arg-Gly-Asp (RGD) for active targeting to αvβ3 integrin, (ii) a hydrophilic motif: D6 for enhancing the solubility in aqueous solution, (iii) a tailoring motif: Gly-Pro-Ala (GPA) for specific recognize and cleavage by FAP-α, (iv) a self-assembly motif: TPE-Phe-Phe (TPE-FF) for in vivo self-assembly with turn-on fluorescence. It is hypothesized that once the peptide-TPE probe active targeting onto the αvβ3 of the pancreatic cancer cells (Miapaca-2 cell line), the membrane protein FAP-α will recognize and tailor the probe between Pro (P) and Ala (A) of the tailoring motif. Then, the self-assembly motif with different peptide sequence modified TPE will leading to in situ self-assembly into nanoarchitectures with dramatically enhanced fluorescence according to the RIR mechanism.

|
Download:
|
| Fig. 1. Chemical structure and modular design of peptide-TPE conjugate (T-FF-P). | |
The azide-modified peptide (N3-GGPG, N3-FFPG, N3-LVFFAPG, N3-FFGPAD6RGD and N3-GGGPAD6RGD) were synthesized by standard solid-phase peptide synthesis methods using Fomc-coupling chemistry [24, 25]. It was further purified by HPLC. Azide-modified peptide and alkyne-modified TPE were coupled by Cu(I)-catalyzed "click" reaction [26-28] using CuSO4/sodium ascorbate as the catalyst in dimethyl sulfoxide (DMSO)/water (1:8, v/v) mixture solvent. Finally, all the self-assembly motifs with different assembled peptide sequence were obtained (Table 1), respectively named as T-OH (TPE-(COOH)2), T-GG (TPE-(GGPG)2), T-FF (TPE-(FFPG)2) and T-LVFF (TPE-(LVFFAPG)2). Additionally, the peptide-TPE probe T-FF-P (TPE-(FFGPAD6RGD)2) and the control probe T-GG-P (TPE-(GGGPAD6RGD)2) were both synthesized accordingly. The synthesized procedure and chemical characterizations can be found in Supporting information (Figs. S1-S11).
|
|
Table 1 Chemical structures of self-assembled peptide-TPE residues. |
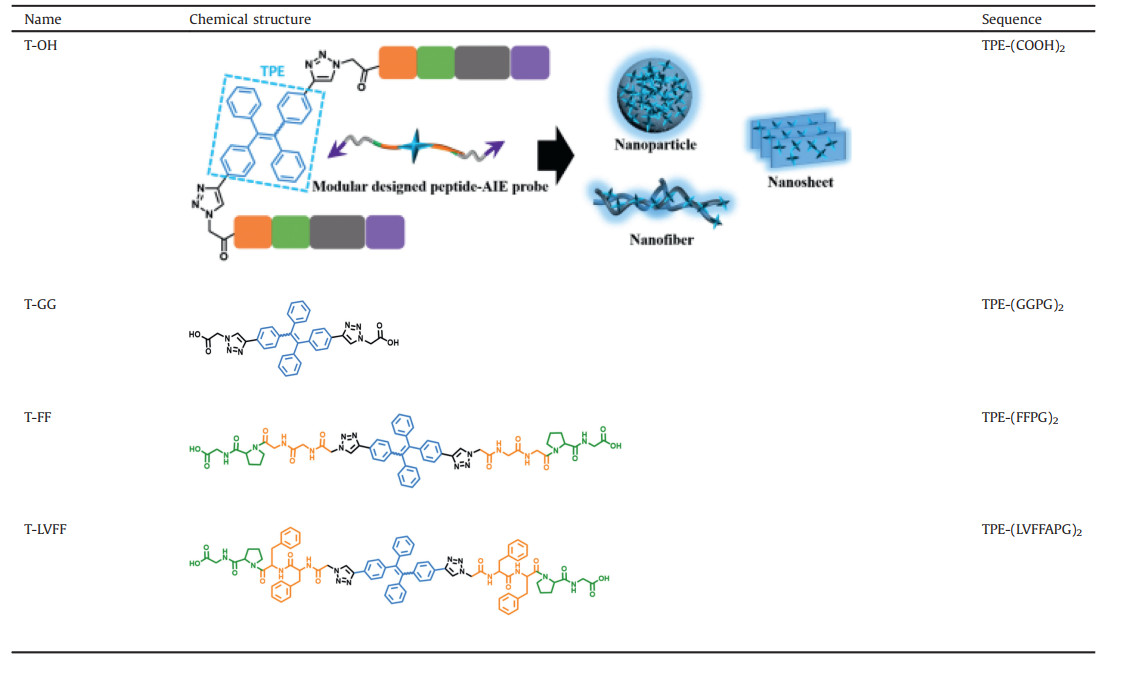
{kind=link}
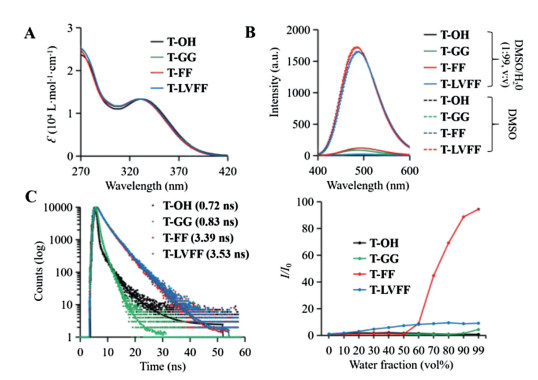
After synthesis of the molecules, the optical properties of four residues were investigated. The UV spectra of T-OH, T-GG, T-FF and T-LVFF in DMSO were almost identical, with absorption peak at 330 nm (Fig. 2A), which mean that the peptide modification did not changed the absorption of TPE. Next, we evaluated the fluorescence properties of the four residues in DMSO/H2O (1:99, v/v) mixture solution compared to the spectra in pure DMSO (Fig. 2B). As known, all these four residues can have a good solubility in DMSO, which usually acted as monomers. The spectra in DMSO (dotted lines) of these molecules had a low fluorescence intensity. Once placed in DMSO/H2O (1:99, v/v) mixture solution (full lines), the well assembled two molecules: T-FF and T-LVFF obviously exhibited an enhanced fluorescence, while, the T-OH and T-GG had no significant change compared with that of in DMSO. The hydrophobicity of the amino acids were the key factors and the aggregated state also impacted the RIR induced emission enhancement. For further demonstration, the time-resolved fluorescence spectra were carried out to verify our conjecture (Fig. 2C). The fluorescent lifetimes of T-FF and T-LVFF are 3.39 ns and 3.53 ns, respectively, which far over the fluorescent lifetimes of T-OH (0.72 ns) and T-GG (0.83 ns). The results indicated that the forbidden nonradiative decay pathway in T-FF and T-LVFF prolonged the fluorescence lifetimes, which may due to the assembly in mixture solution. The critical micelle concentration (CMC) of T-GG (22.4 μmol/L) and T-FF (7.9 μmol/L) both confirmed the hypotheses above (Fig. S12 in Supporting information). The AIE behaviors of these four residues were further studied by recording the fluorescence turn-on ratios (I/I0) in Fig. 2D and Figs. S13-S16 (Supporting information). When the water fractions were below 50 %, T-FF remained nonfluorescent. However, with the water fraction increasing, T-FF became highly emissive, a characteristic performance of AIEgens. However, T-OH and T-GG did not show obvious AIE properties upon increased water fraction, due to the lack of hydrophobicity and π-π interaction of side chains of molecules. Meanwhile, the T-FF not only exhibited stronger intermolecular interactions, but also formed ordered fibril structures dependent on the β-sheet assembly, resulting in obvious AIE properties. While, T-LVFF shows strong fluorescence intensity at low water content due to the strong hydrophobic peptide sequence, which was not benefit for sensitive bioimaging in biological condition.

|
Download:
|
| Fig. 2. Photophysical properties of self-assembled residues of T-OH, T-GG, T-FF and T-LVFF. (A) UV–vis absorption spectra in DMSO. (B) FL spectra of assemblies in mixture solution of DMSO/H2O (1:99, v/v) and monomers in DMSO. (C) Time-resolved fluorescence spectra. (D) Relative variations of the fluorescent intensity I/I0 (where I0 is defined as the baseline fluorescent intensity in pure DMSO) in different mixture solutions of DMSO/H2O. λex: 370 nm, λem: 480 nm, concentration: 10 μmol/L. | |
{kind=link}
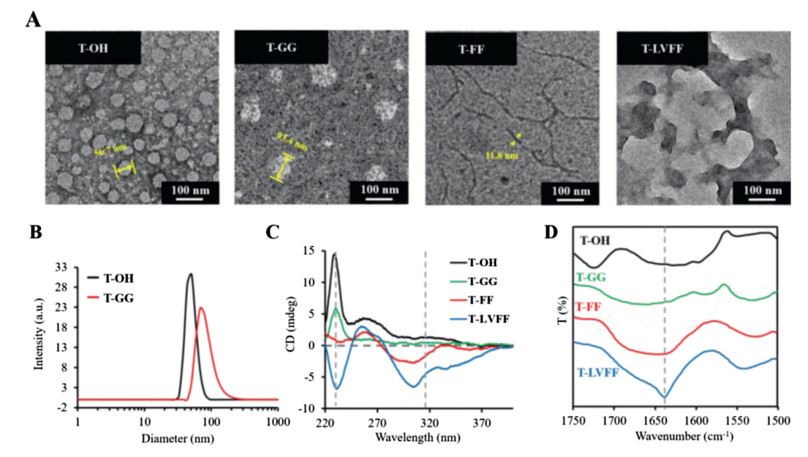
The assembled morphology of the different peptide-TPE residues were validated accordingly (Fig. 3A). The transmission electron microscopy (TEM) observations indicated that both the T-OH and T-GG aggregates formed nanoparticles with a diameter around 56.7 nm and 93.4 nm, which was well matched the size distribution from dynamic light scattering (DLS) in Fig. 3B. It has been reported that FF with an aromatic capping group and LVFFA usually self-assembled into nanomaterials based on multiple weak bond interactions induced β-sheets assembly [29]. As shown in Fig. 3A, the T-FF formed typical nanofibers and T-LVFF forms lamellar nanosheets due to hydrophobic interaction, π-π stacking and hydrogen bonding based on peptides.

|
Download:
|
| Fig. 3. Morphology and spectra of the nano-assemblies. (A) TEM images of assembled morphology of T-OH, T-GG, T-FF and T-LVFF. (B) Dynamic light scattering (DLS) of assembled T−OH and T-GG nanoparticles. (C) Circular dichroism (CD) spectra. (D) Fourier transform infrared (FTIR) spectra of four residues in aqueous solution. Concentration: 50 μmol/L. | |
{kind=link}
To further study the self-assembly behavior of the assemblies, the circular dichroism (CD) and Fourier transform infrared (FTIR) spectra were carried out. As shown in Fig. 3C, the four residues revealed a strong cotton effect band approximate at 230 nm, which was due to the aromatic π-π effects of TPE and phenylalanine [30]. In addition, CD spectra of T-FF and T-LVFF showed a cotton effect signals around 320 nm, indicating strong π-π stacking of TPE chromophores in water [31]. All the results indicated that peptide modification strongly induced intermolecular interactions, which would sequently effect the optical property of TPE. As known, FTIR is a powerful tool for investigating intermolecular interactions. As shown in Fig. 3D, the bands of T-LVFF and T-FF in the amide I region were significantly shifted to lower energy regions relative to T-GG and T-OH, suggesting that the assembles form of T-LVFF and T-FF existed obviously hydrogen bonding based on modified peptides. Meantime, the stronger intermolecular interactions of T-LVFF showed a peak around 1640 cm−1, which can be identified as antiparallel β-sheet assembly.
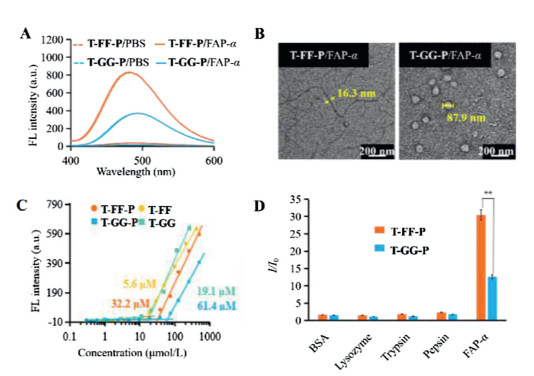
Based on the assembled behavior and turn-on fluorescence property in aqueous solution, T-FF-P was chosen as the suitable candidate bioactivated in vivo assembly (BIVA) probe for cell imaging. Meantime, the T-GG-P was set as control probe modified with no obvious assembly inducing peptide. To verify the turn-on behavior of T-FF-P and T-GG-P, we performed enzymatic assays with recombinant human fibroblast activation protein (FAP-α) in vitro. Mixtures of BIVA probes (20 μmol/L) and FAP-α (100 pmol/L) were prepared and incubated in PBS buffer (pH 7.5) at 37℃ for 3 h and then the fluorescence spectra were obtained in the range from 400 nm to 600 nm. As shown in Fig. 4A, the water-soluble T-FF-P and T-GG-P do not emit fluorescence in PBS buffer, but switch to fluoresce once incubated with FAP-α (100 pmol/L). LC-MS spectra of the T-FF-P probes before and after triggered by FAP-α in buffer was carried out (Fig. S17 in Supporting information). As a result, after FAP-α tailoring, the original T-FF-P peak with a retention time of 25.1 min disappeared, sequently, two peaks at 11.1 min and 20.9 min were newly appeared which was identified by MOLDI-TOF, which were corresponding to peptide residues of ADDDDDDRGD and T-FF. Both the probes were in a random secondary structure from CD spectra (Fig. S18 in Supporting information). The different assembled residues of these two probes acted obviously different light-on property, which may due to the assembled architectures differed the intramolecular rotation restriction of TPE. The FF peptide exhibited more effective π-π stacking between the peptide and TPE than that of GG peptide, resulting in enhanced fluorescence. Next, the assembled morphology of nanostructures of T-FF-P and T-GG-P after FAP-α co-incubation in buffer were observed by TEM images (Fig. 4B). It was found that the T-FF-P residues (T-FF-P) were able to form filamentous nanostructures with a width around 16.3 nm. In sharp comparison, the T-GG-P residues (T-GG) formed nanoparticles with a diameter around 80.4 nm, which was matched with the size measured by DLS (87.9 nm) (Fig. S19 in Supporting information). The difference between T-FF-P and T-GG-P residues assemblies in morphology revealed their different intrinsic molecular arrangements. To quantify the concentration-dependent aggregation behavior of T-FF-P and T-GG-P, we measured their critical micelle concentration (CMC) values. As displayed in Fig. 4C, the CMC of T-FF-P was 32.2 μmol/L, which was much lower than that of T-GG-P (61.4 μmol/L). Their residues in buffer were relative lower than that of in water. The results suggested that FF as the self-assembly-aiding unit, not only modulated the intermolecular interactions but also increased the hydrophobicity resulting in CMC reduction. To further investigate the specific recognition ability of the probes to FAP-α, T-FF-P and T-GG-P were tested by different proteins, such as FAP-α, bovine serum albumin (BSA), lysozyme, trypsin and pepsin, under identical conditions. As shown in Fig. 4D, FAP-α groups displayed around 14–16 and 4−6-fold higher in I/I0 than the other groups. This substantiated that T-FF-P and T-GG-P certainly highly sensitive and specific to FAP-α in vitro.

|
Download:
|
| Fig. 4. Enzyme specific tailoring induced assembly and fluorescent light-up. (A) FL turn-on response of T-FF-P and T-GG-P (20 μmol/L) with FAP-α (100 pmol/L). (B) TEM images of T-FF-P and T-GG-P after treatment with FAP-α. (C) The critical micelle concentration (CMC) of T-FF-P and T-GG-P, as well as their tailored residues T-FF and T-GG in buffer. (D) FAP-α specificity responsiveness. Plot of I/I0 vs. different proteins, I and I0 are the FL intensities at the protein concentration of 100 and 0 pmol/L, respectively. Statistical significance is assessed by Student's test, **P < 0.01. | |
{kind=link}
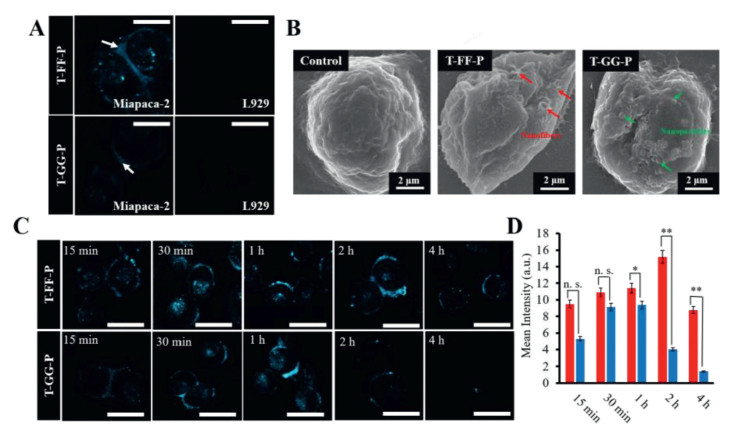
FAP-α is identified as a diagnostic biomarker to distinguish the tumor activated fibroblasts from the normal fibroblasts, which is highly expressed in pancreatic tumor cells (e.g., Miapaca-2 cells). After demonstrating the BIVA probes' excellent sensitivity and specificity to FAP-α in buffer, we selected Miapaca-2 (FAP-α positive expressed tumor cell) and L929 (FAP-α negative expressed normal cell) to evaluate the bioimaging property. First of all, we evaluated the cytotoxicity of T-FF-P and T-GG-P to L929 and Miapaca-2 cells using CCK-8 assay. After 24 h incubation, there had no significant cytotoxicity to cells when the concentration up to 20 μmol/L (Fig. S20 in Supporting information). Then, the BIVA probes were respectively co-incubated with Miapaca-2 cells and L929 cells under a concentration of 10 μmol/L at 37℃ for 30 min (Fig. 5A). Miapaca-2 and L929 cells treated with PBS as contral (Fig. S21 in Supporting information). As expected, negligible fluorescence signal was observed in both the T-FF-P-treated and T-GG-P-treated L929 cells. On the contrary, T-FF-P and T-GG-P co-incubated with Miapaca-2 cells had a significant signal difference, which increased the fluorescence signal on the membrane of cell after treatment of T-FF-P. Moreover, the fluorescence signal of T-GG-P was much less than that of T-FF-P. These results indicated that T-FF-P can specifically respond to FAP-α overexpressed Miapaca-2 cells and T-FF-P was much more sensitive than T-GG-P for cell imaging. To further validating if the assembled structures were different leading to the bioimaging sensitivity, we further performed scanning electron microscope (SEM) imaging (Fig. 5B). Comparing with the control cell surface morphology, the T-FF-P treated cell appeared nanofiber structures (red arrows) and the T-GG-P ones appeared nanoparticles (green arrows), which was co-coordinated with the results in solution. Thus, the different nanostructure assembled on cells and sequently differed the enhanced fluorescence both contributed to the cell imaging sensitivity. Moreover, real-time fluorescence imaging profiles of Miapaca-2 cells with these two BIVA probes were carried out. As time goes on, rarely fluorescence signal of T-GG-P was observed at first 15 min, highest fluorescence signal was captured at 1 h and almost disappeared after 4 h. Conversely, long-term cell surface fluorescence of T-FF-P was observed begin in 15 min, reached to the lightest signal up to 2 h and lasted up to 4 h (Fig. 5C). The signal at 2 h of T-FF-P was 3.8-fold higher than that of T-GG-P (Fig. 5D). It was indicated the nanofibrous structure strongly enhanced the fluorescence signal and lasted the signal retention providing a better imaging window.

|
Download:
|
| Fig. 5. Bioactivated in vivo assembly (BIVA) peptide-TPE probe for pancreatic tumor cell imaging. (A) Confocal images of Miapaca-2 and L929 cells treated with probes. (B) SEM images of cell surfaces of Miapaca-2 treated with T-FF-P and T-GG-P and untreated Miapaca-2 as a control. (C) Confocal images of time-dependent monitoring of Miapaca-2 cells treated with the probes (20 μmol/L) for 15 min followed by washing with PBS and replacing the medium and a further incubation up to 4 h. (D) Quantitative analysis of mean fluorescence intensity on the Miapaca-2 cells from confocal images with a timescale range of 15 min-4 h. Statistical significance is assessed by Student's test, *P < 0.05, ** P < 0.01, n.s.P > 0.05. Scale bar of all confocal images: 50 μm. | |
{kind=link}
In conclusion, we have reported a FAP-α specific BIVA peptide-TPE probe for enhanced pancreatic tumor cell imaging. The peptide sequence coordinated the assembled behavior of TPE through the assembled residues, inducing a defined nano-structure with modulated intermolecular interactions. In addition, the coupling of the tailoring motif, hydrophilic motif and targeting motif on the assembled residue endowed the obtained BIVA probe with active targeting and tailoring induced retention effect, light-up property of AIE signal in aqueous solution, and nanostructure modulated biofunctions [32-34]. After optimization, the FF peptide exhibited a best balance of solubility of the probe and the nanofibrous assembled structure, yielding close and tight intermolecular steric interactions to restrict the intramolecular motions of TPE for excellent signal output [35, 36]. Finally, the designed BIVA probe obtained a highly sensitive and specific property for bioimaging to pancreatic tumor cells. We believed that the peptides enable to modulating the AIE assembly, sequently endowed the probe better photophysical property and biomedical effect base on certain nanostructures.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Key R & D Program of China (No. 2018YFE0205400), National Natural Science Foundation of China (Nos. 31671028 and 51873045), Youth Innovation Promotion CAS (No. 2017053). Thanks Prof. Ben Zhong Tang for supplying the alkynylated tetraphenylethylene (TPE) molecules.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.01.007.
| [1] |
Y. Hong, J.W. Lam, B.Z. Tang, Chem. Commun. (2009) 4332-4353. |
| [2] |
R. Hu, N.L.C. Leung, B.Z. Tang, Chem. Soc. Rev. 43 (2014) 4494-4562. DOI:10.1039/C4CS00044G |
| [3] |
J. Zhao, D. Yang, Y. Zhao, et al., Angew. Chem. Inter. Ed. 53 (2014) 6632-6636. DOI:10.1002/anie.201402169 |
| [4] |
K.C. Chong, F. Hu, B. Liu, Mater. Chem. Frontiers 3 (2019) 12-24. DOI:10.1039/C8QM00383A |
| [5] |
F. Hu, S. Xu, B. Liu, Adv. Mater. 30 (2018) e1801350. DOI:10.1002/adma.201801350 |
| [6] |
R.V. Ulijn, A.M. Smith, Chem. Soc. Rev. 37 (2008) 664-675. DOI:10.1039/b609047h |
| [7] |
E. De Santis, M.G. Ryadnov, Chem. Soc. Rev. 44 (2015) 8288-8300. DOI:10.1039/C5CS00470E |
| [8] |
J. Wang, K. Liu, R. Xing, X. Yan, Chem. Soc. Rev. 45 (2016) 5589-5604. DOI:10.1039/C6CS00176A |
| [9] |
G. Wei, Z. Su, N.P. Reynolds, et al., Chem. Soc. Rev. 46 (2017) 4661-4708. DOI:10.1039/C6CS00542J |
| [10] |
G.B. Qi, Y.J. Gao, L. Wang, H. Wang, Adv. Mater. 30 (2018) e1703444. DOI:10.1002/adma.201703444 |
| [11] |
A. Han, H. Wang, R.T.K. Kwok, Anal. Chem. 88 (2016) 3872. DOI:10.1021/acs.analchem.6b00023 |
| [12] |
L.L. Li, Z.Y. Qiao, L. Wang, H. Wang, Adv. Mater. 31 (2019) e1804971. DOI:10.1002/adma.201804971 |
| [13] |
D.B. Cheng, D. Wang, Y.J. Gao, et al., J. Am. Chem. Soc. 141 (2019) 4406-4411. DOI:10.1021/jacs.8b13512 |
| [14] |
X.X. Zhao, L.L. Li, Y. Zhao, et al., Angew. Chem. Int. Ed. 58 (2019) 15287-15294. DOI:10.1002/anie.201908185 |
| [15] |
X. Ni, X.Y. Zhang, Nano Lett. 19 (2019) 318-330. DOI:10.1021/acs.nanolett.8b03936 |
| [16] |
X. Zhao, L. Li, H. Wang, Chin. Sci. Bull. 63 (2018) 1088-1094. DOI:10.1360/N972018-00148 |
| [17] |
L.L. Li, S.L. Qiao, W.J. Liu, et al., Nat. Commun. 8 (2017) 1276. DOI:10.1038/s41467-017-01296-8 |
| [18] |
K.F. Kelly, PNAS 123 (2000) 342-345. |
| [19] |
F. Hu, D. Mao, Kenry, et al., Angew. Chem. Int. Ed. 57 (2018) 10182-10186. DOI:10.1002/anie.201805446 |
| [20] |
M. Duo, H. Fang, Kenry, et al., Adv. Mater. 30 (2018) 1706831. DOI:10.1002/adma.201706831 |
| [21] |
Y. Zhang, P. Huang, J. Guo, Adv. Mater. 32 (2020) 23. |
| [22] |
Z. Zhao, X. Zheng, L. Du, et al., Nat. Commun. 10 (2019) 2952. DOI:10.1038/s41467-019-10818-5 |
| [23] |
R.L. Noble, G.B. Fields, Int. J. Pept. Protein Res. 35 (1990) 161-214. |
| [24] |
M. Olaru, A. Mischin, L.A. Malaspina, Angew. Chem. Int. Ed. 59 (2020) 232-237. DOI:10.1002/anie.201911516 |
| [25] |
R. Behrendt, P. White, J. Offer, J. Pept. Sci. 22 (2016) 4-27. DOI:10.1002/psc.2836 |
| [26] |
J.H. Cho, F. Amblard, R.F. Schinazi, Chem. Rev. 109 (2009) 4207-4220. DOI:10.1021/cr9001462 |
| [27] |
H. Shi, R.T. Kwok, J. Liu, et al., J. Am. Chem. Soc. 134 (2012) 17972-17981. DOI:10.1021/ja3064588 |
| [28] |
Y. Yuan, C.J. Zhang, M. Gao, et al., Angew. Chem. Int. Ed. 127 (2015) 1800-1806. DOI:10.1002/ange.201408476 |
| [29] |
L.L. Li, Z.Y. Qiao, L. Wang, H. Wang, Adv. Mater. 31 (2019) e1804971. DOI:10.1002/adma.201804971 |
| [30] |
N.R. Lee, C.J. Bowerman, B.L. Nilsson, Biomacromolecules 14 (2013) 3267-3277. DOI:10.1021/bm400876s |
| [31] |
N.T. Chu, R.D. Chakravarthy, N.C. Shih, et al., RSC Adv. 8 (2018) 20922-20927. DOI:10.1039/C8RA02296H |
| [32] |
Y. Cui, R. Zhang, L. Yang, Chin. Chem. Lett. 30 (2019) 1078-1082. DOI:10.1016/j.cclet.2018.10.017 |
| [33] |
J.Y. Gong, P.F. Wei, Y.A. Su, Y. Li, Chin. Chem. Lett. 29 (2018) 79-82. |
| [34] |
S. Zhang, J.M. Yan, A.J. Qin, Chin. Chem. Lett. 24 (2013) 668-672. DOI:10.1016/j.cclet.2013.05.014 |
| [35] |
C.Y. Yu, C.C. Hsu, Polymer 137 (2018) 30-37. DOI:10.1016/j.polymer.2018.01.002 |
| [36] |
H. Zhou, S. Li, X. Zeng, Chin. Chem. Lett. 31 (2020) 1382-1386. DOI:10.1016/j.cclet.2020.04.030 |