 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Pesticide and Chemical Biology, Ministry of Education, Hubei International Scientific and Technological Cooperation Base of Pesticide and Green Synthesis, International Joint Research Center for Intelligent Biosensing Technology and Health, College of Chemistry, Central China Normal University, Wuhan 430079, China;
c CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
A deep understanding of photophysical and photochemical mechanisms is critical for the rational design of optoelectronic materials [1-3]. Most of these existing mechanisms can be broadly classified into two categories: (1) energy transfer and (2) charge/electron transfer [4]. The applications of the respective mechanisms have enabled many useful technologies [5]. Yet, the combined effects of both energy transfer and electron transfer in a single donor-acceptor system received far less attention. Understanding the collective effects of energy and electron transfers at the molecular level, however, could pave new avenues for developing unprecedented functional materials.
To date, energy transfer processes have been thoroughly studied in various materials systems [6, 7]. The mechanisms of energy transfer include both Förster resonance energy transfer (FRET) [8] and Dexter energy transfer (DET) [9]. FRET takes place mainly via dipole-dipole interactions between an energy donor (D) and acceptor (A). The efficiency of FRET critically depends on the distance and orientation between the donor and the acceptor, as well as the spectral overlap between the donor's emission and the acceptor's absorption spectra [10]. DET occurs via a double electron-transfer process: Upon photoexcitation of the donor, one electron transfers from the donor LUMO to the acceptor LUMO; the other from the acceptor HOMO to the donor HOMO. The efficiency of DET strongly depends on the donor-acceptor distance and the overlap between donor and acceptor orbitals [10]. FRET usually takes place from 10 Å to 100 Å, while DET typically occurs within 10 Å [11]. Notably, both FRET and DET have enabled numerous biophysical and biochemical applications, such as detecting various biomolecules and ions [12], studying protein conformational changes [13], sequencing DNAs [14], monitoring polymerase chain reactions [15], constructed high-resolution microenvironment mapping [16] and developing fluorogenic dyes [17].
Photoinduced electron transfer (PET) is another fundamental photophysical mechanism [18]. During PET, a single electron moves from the electron donor to the acceptor, leading to the formation of charge-separated bi-radicals, which are highly reactive and non-emissive [19]. PET strongly depends on the energy levels of the donor and acceptor frontier molecular orbitals, the distance between the electron donor and acceptor, and solvent polarity [20]. PET also leads to the creations of many fluorescent functional materials. For example, de Silva pioneered the field of molecular logic gates [21], many of which utilize PET to modulate fluorescent output [22]. Nagano et al. developed highly sensitive nitric oxide [23] and nitrative probes [24]. Our group showed that PET fluorophores allowed wash-free bioimaging of lipid droplets and mitochondria, and provided a new route for constructing AIEgens [20].
Currently, most existing photosystems employ either energy transfer or electron transfer [25]. However, research on the combined effect of both energy transfer and electron transfer in a single donor-acceptor dyad remains sporadic, especially in purely organic compounds [26-29]. In this manuscript, we reported an energy transfer followed by electron transfer (ETET) mechanism in an organic dyad TPE-NBD (Scheme 1a). With the help of fluorescence output to probe the short-lived excited-state dynamics, our combined experimental and computational studies provided unambiguous evidence on the polarity dependent ETET processes. We expect that ETET would empower many useful applications, such as bright multicolour fluorescence imaging, sensing, as well as photodynamic and photothermal therapies.

|
Download:
|
| Scheme 1. (a) Schematic illustration of the ETET mechanism. (b) Chemical structures of TPE, NBD, TPE&NBD, and TPE-NBD. | |
{kind=link}
To investigate the combined effect of energy transfer and electron transfer, we prepared a molecular dyad TPE-NBD along with three reference compounds: TPE, NBD, and TPE&NBD (the mixture of TPE and NBD of equal concentrations (Scheme 1b)). The synthesis details can be found in Supporting information. During sample preparation, we kept the concentrations of all compounds low at 10 μmol/L and conducted additional tests to ensure no aggregate formations (Fig. S3 in Supporting information). Subsequently, we measured the UV–vis absorption spectra of these compounds (Fig. 1a). The first absorption band of TPE peaked at 307 nm in dioxane, while NBD exhibited two absorption bands peaked at 452 nm and 322 nm, respectively. As expected, TPE&NBD showed a superimposed UV–vis spectrum of TPE and NBD. Most interestingly, the UV–vis spectrum of TPE-NBD is (almost) identical to that of TPE&NBD (considering experimental errors). The UV–vis spectra in other solvents showed the same pattern (Fig. S1 in Supporting information). These identical spectra between TPE&NBD and TPE-NBD led us to conclude that the TPE and NBD fragments in TPE-NBD are spectroscopically independent units in the ground state.

|
Download:
|
| Fig. 1. (a) UV–vis absorption spectra of TPE, NBD, TPE&NBD and TPE-NBD in dioxane. Fluorescence spectra of TPE, NBD, TPE&NBD and TPE-NBD in (b) toluene excited at 310 nm and (c) DMSO excited at 300 nm. (d) Fluorescence spectra of TPE-NBD in various solvents excited at the respective peak UV–vis absorption wavelength. [TPE]=[NBD]=[TPE&NBD]=[TPE-NBD]=10 μmol/L; all tests are done at room temperature. | |
{kind=link}
To probe the potential energy transfer between TPE and NBD, we measured the emission spectra of all four compounds by exciting them at 310 nm (around the peak UV–vis absorption wavelength of the TPE fragment; Fig. 1b). Compound TPE is not fluorescent, due to significant molecular motions and associated non-radiative decays in the excited state [30, 31]. Compounds NBD and TPE&NBD emit moderately bright fluorescence. In TPE&NBD, the intermolecular distance between TPE and NBD seems too large for efficient intermolecular energy transfer to occur; besides, TPE in TPE&NBD does not emit. It is thus not surprising that NBD and TPE&NBD demonstrate similar fluorescence features as NBD is the only fluorescent component. In contrast, the emission intensities of TPE-NBD are greatly intensified by ~3 times in comparison to those of NBD and TPE&NBD. The enhanced emission of TPE-NBD indicated efficient energy transfer from the TPE moiety to the closely linked NBD fragment. The emission spectra in other non-polar solvents also revealed such an energy transfer process (Fig. S2 in Supporting information). Interestingly, these results show that during the energy transfer process, the energy donor (such as TPE) does not have to be emissive.
Nonetheless, as the solvent polarity increased, the emission intensity of TPE-NBD experienced a substantial reduction (Table S1 in Supporting information). For example, when we excited all four compounds at 300 nm in DMSO, the fluorescence of TPE-NBD became much weaker than that of NBD and TPE&NBD (Fig. 1c). These results show that in addition to energy transfer, a quenching mechanism becomes dominant in polar solvents for TPE-NBD. In addition, considering that this quenching process is notable only in TPE-NBD (but not NBD), we conclude that this fluorescence quenching is related to the interactions between TPE and NBD. Finally, a comparison of the emission intensities between TPE-NBD (weak) and TPE&NBD (strong) showed that this quenching process also depends on the distance between TPE and NBD. That is, a close distance leads to more effective quenching in polar solvents.
To shed light on the underlying mechanism, we directly excited the NBD fragment of TPE-NBD at ~450 nm. In this way, we avoided energy transfer from TPE to NBD. The results show that the emission of the NBD fragments still strongly decreases as the solvent polarity increases (Fig. 1d and Fig. S4 in Supporting information). Taking the above observations into account, we hypothesized that the quenching process was a consequence of photoinduced electron transfer between TPE and NBD. That is, energy transfer followed by electron transfer (ETET) in TPE-NBD takes place in polar solvents.
We also collected the fluorescence excitation spectra and measured the fluorescence lifetime of TPE-NBD in different solvents (Fig. S5 and Table S2 in Supporting information). In agreement with the emission data, the fluorescence excitation spectra become weak with increasing solvent polarity. Similarly, the fluorescence lifetime of TPE-NBD dropped from 9.43 ns in toluene to nearly 0 in DMSO. These data are consistent with the polarity dependence of PET. That is, high-polarity solvents enhance PET [20].
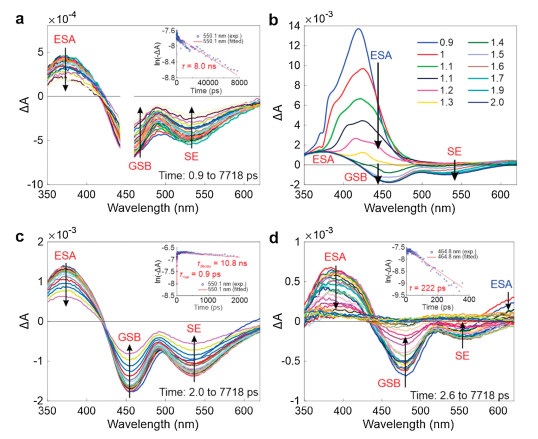
To gain further insights into the ETET mechanism, we collected transient absorption spectra (TAS) of TPE-NBD. We firstly conducted the experiments in dioxane, where TPE-NBD is highly emissive. Upon direct excitation of the NBD fragment at 450 nm, we observed an excited state absorption (ESA) band from ~350 nm to ~420 nm, a ground state bleaching (GSB) band from ~420 nm to ~490 nm, and a stimulated emission (SE) band from ~490 nm to ~620 nm (Fig. 2a). In particular, the GSB and SE bands perfectly match the steady-state UV–vis absorption and emission spectra of TPE-NBD (or its NBD fragment), respectively. We also noted that ESA/GBS/SE bands of the NBD fragment have a long lifetime of ~8 ns, in good agreement with the bright emission of TPE-NBD (Fig. 2a inset and Fig. S6 in Supporting information).

|
Download:
|
| Fig. 2. (a) Transient absorption spectra of TPE-NBD in dioxane, upon excitation at 450 nm; the inset shows the corresponding decay dynamics at 550.1 nm and the single-exponential fitting. Transient absorption spectra of TPE-NBD in dioxane (b) within 2 ps and (c) from 2 ps to 7718 ps, upon excitation at 340 nm; the inset in (c) shows the corresponding decay dynamics and the exponential fitting at 550.1 nm. (d) Transient absorption spectra of TPE-NBD in DMSO from 2.6 ps to 7718 ps, upon excitation at 340 nm; the inset shows the corresponding decay dynamics and the single-exponential fitting at 464.8 nm. | |
{kind=link}
When exciting TPE-NBD at 340 nm (by aiming at the TPE fragment), energy transfer from TPE to NBD was observed (Figs. 2b and c). We noted a new ESA band peaked at ~420 nm and attributed it to the TPE fragment, due to its similarity to previously reported data of TPE [30]. The ESA of TPE rapidly decayed with a lifetime of ~1 ps. Following this decay, we noticed the formation of the ESA/GSB/SE bands of the NBD fragment, indicating energy transfer from the TPE fragment to the NBD moiety in TPE-NBD. Indeed, by probing ΔA dynamics at 550 nm in the SE band, we observed a rapid intensification with a lifetime of 0.9 ps, followed by a slow decay (with a lifetime of 10.8 ns; Fig. 2c inset). Similar slow decays were also noted in the ESA and GSB bands of the NBD moiety (Fig. S7 in Supporting information).
We next performed the TAS experiments in DMSO, where TPE-NBD became non-emissive. Upon exciting TPE-NBD at 340 nm, we again observed the ESA band of the TPE moiety (Fig. S8 in Supporting information). Interestingly, the peak of this ESA band continuously evolved, suggesting significant conformational changes of TPE in polar solvents. This ESA band also experiences a rapid decay within ~2 ps, followed by the formation of the ESA/GSB/SE bands of the NBD moiety, indicating energy transfer from TPE to NBD (Fig. 2d).
Notably, the de-excitation rate of the NBD fragment is significantly enhanced in DMSO. For example, the decay lifetime of NBD is shortened to 222 ps in DMSO (vs. ~10 ns in dioxane), following the nearly zero quantum yield of TPE-NBD in polar solvents (Fig. 2d inset and Fig. S9 in Supporting information). The rapid decay of the NBD fragment suggests the presence of a significant non-radiative process, i.e., photoinduced electron transfer (PET). However, in our TAS experiments, we did not directly observe the formation of the electron-transfer (ET) state, probably owing to the limited wavelength range covered in our TAS experiments.
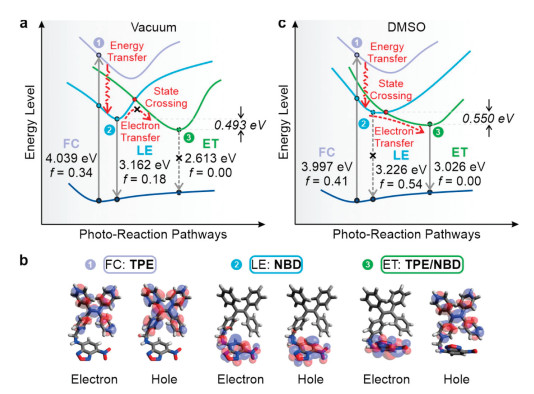
To reveal the fluorescence quenching mechanism, we performed detailed excited-state calculations of TPE-NBD at the M06−2X/def2SVP level of theory in vacuo (Figs. 3a and b, and Fig. S10 in Supporting information). Our computational results showed that by exciting TPE-NBD at short wavelengths, the energy is largely absorbed by the TPE fragment [in the Franck Condon (FC) state; highlighted in light purple], and then quickly transfer to the NBD fragment [the locally-excited (LE) state; highlighted in blue], as the distance between TPE and NBD moieties is small (7.066 Å; Fig. S11 in Supporting information) in TPE-NBD. The LE state is highly emissive with a relatively large oscillator strength (f=0.18), corresponding to the emission from the NBD moiety. Moreover, on the S1 potential energy surface, a low-lying dark electron-transfer state (f=0) exists (the ET state; highlighted in green). This ET state corresponds to electron transfer from the TPE to NBD fragments in TPE-NBD, thus corroborating our hypothesis of PET as the quenching mechanism. Besides, this ET state is more stable than the LE state by 0.493 eV, suggesting a potential PET quenching even in non-polar environments. Nonetheless, given that TPE-NBD is highly emissive in non-polar solvents, the energy barrier to enter this dark ET state via state-crossing should be high. In other words, only energy transfer occurs in TPE-NBD, while electron transfer is constrained at a minimal level in non-polar environments.

|
Download:
|
| Fig. 3. Schematic illustration of the state-crossing from a locally excited to an electron transfer state and calculated excitation/de-excitation energy (as well as oscillator strength) of TPE-NBD (a) in vacuo and (c) in DMSO. (b) Optimized molecular structures of TPE-NBD with the corresponding electron and hole distributions in vacuo. The energy levels in (a) and (c) were not drawn to scale for clarity. | |
{kind=link}
It is of note that that the energy transfer in TPE-NBD was Dexter energy transfer (DET), because the distance between TPE and NBD fragments is less than 10 Å. Moreover, between two types of energy transfer, the efficiency of FRET depends on the quantum yield of the energy donor (TPE). When the energy donor is non-emissive (with a quantum yield of 0), the rate of FRET becomes negligible [32]. In contrast, DET does not depend on the donor quantum yield, but only on the overlap between the normalized donor emission and normalized acceptor absorption spectra. Accordingly, even though TPE is not emissive, efficient energy transfer could still take place in TPE-NBD via DET.
As solvent polarity increases, the highly polar ET state should become much more stable resulting in a lower energy barrier for the state-crossing to occur. This is due to strong dipole-dipole and induced dipole interactions with polar solvent molecules. Indeed, our calculations show that the energy gap between LE and ET states enlarged from 0.493 eV in vacuo to 0.550 eV in DMSO (Fig. 3c). The further stabilized ET state could substantially activate PET, thus significantly quenching the fluorescence of TPE-NBD in polar solvents. These theoretical predictions are fully consistent with experimental observations.
Interestingly, on the basis that the ET state is more stable than the LE state in all environments, our calculations predicted that compound NBD (without PET) should be brighter than TPE-NBD (with different degrees of PET as solvent polarity increases) in the absence of energy transfer (by directly exciting the NBD moieties). Inspired by this prediction, we normalized the emission intensities of TPE-NBD and NBD to the UV–vis absorbance at the excitation wavelength (to eliminate concentration dependence) and compared the resulted intensities (Fig. S12 in Supporting information). Our results show that TPE-NBD is indeed less emissive than NBD, confirming the presence of a dark state in TPE-NBD. These data unambiguously show that energy transfer followed by electron transfer (ETET) could take place in molecular dyads.
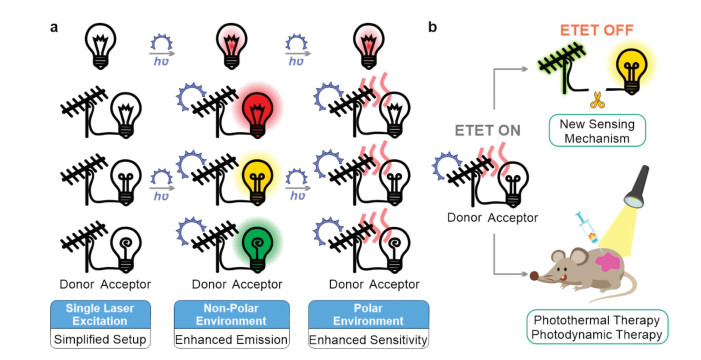
We found that the ETET mechanism was also applicable to many other compounds (Fig. S13 in Supporting information) [33]. We also expect that ETET has a broad range of applications. For example, in multicolour imaging and sensing, several lasers are often required to excite various fluorophores with distinct absorption and emission bands. With ETET, however, only one laser that matches the λabs of the energy donor is needed, which could greatly simplify the optical setup and reduce the cost (Fig. 4a). Moreover, since many fluorophores have a low molar extinction coefficient, connecting an energy donor with a high molar extinction coefficient could greatly boost the brightness of various fluorophores via energy transfer in non-polar solvents. In contrast, in polar solvents, the activation of electron transfer could completely quench the emissions of all dyads. Collectively, ETET could endow these compounds with greatly enhanced environmental sensitivities, which are useful for fluorescent sensing and fluorogenic imaging.

|
Download:
|
| Fig. 4. Schematic illustrations of ETET based applications. (a) Single-laser powered multicolor labelling and sensing applications via ETET; (b) Photothermal/photodynamic therapy and reaction-based sensing applications based on ETET. | |
{kind=link}
Besides, the linker between the donor and acceptor fragments served as one additional reaction site for modulating the ETET process (Fig. 4b). When the donor and the acceptor are very close to each other, both energy transfer and electron transfer could happen in polar solvents, thus quenching the emission. When the linker is cut off via biochemical reactions, ETET becomes inhibited, with the potential turn-on of dual-colour fluorescence (from the donor and acceptor fragments, respectively) for ratiometric imaging. Finally, when ETET is on (with no fluorescence), such dyads could also serve as useful agents for photothermal and photodynamic therapies (Fig. 4b).
In summary, through a detailed experimental and theoretical investigation of the fluorescence characteristics of a molecular dyad TPE-NBD, we revealed a distinct photophysical mechanism: energy transfer followed by electron transfer (ETET). As a result of the energy transfer, this dyad exhibit greatly enhanced brightness in non-polar solvents. In contrast, with the activation of photoinduced electron transfer (after energy transfer) in polar solvents, this dyad becomes non-emissive. We expect that the enhanced environmental sensitivity via the combined effects of both energy and electron transfer endow ETET with a great potential for a wide range of applications, such as in multicolour bioimaging via a single laser excitation, ratiometric biosensing, photodynamic and photothermal therapies.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by Singapore University of Technology and Design (No. T1SRCI17126) and A*STAR under its Advanced Manufacturing and Engineering Program (No. A2083 c0051), the National Natural Science Foundation of China (Nos. 21878286, 21908216), and Dalian Institute of Chemical Physics (Nos. DMTO201603, TMSR201601). The authors are grateful for the computing service of SUTD-MIT IDC and the National Supercomputing Center (Singapore).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.038
| [1] |
S. Berardi, S. Drouet, L. Francàs, et al., Chem. Soc. Rev. 43 (2014) 7501-7519. DOI:10.1039/C3CS60405E |
| [2] |
J. Zhao, Y. Li, G. Yang, et al., Nat. Energy 1 (2016) 1-7. |
| [3] |
L. Zhou, L. Xie, C. Liu, Y. Xiao, Chin. Chem. Lett. 30 (2019) 1799-1808. DOI:10.1016/j.cclet.2019.07.051 |
| [4] |
J.R. Lakowicz, Principles of Fluorescence Spectroscopy. 3rd ed.. New York: Springer, 2006.
|
| [5] |
K.E. Sapsford, L. Berti, I.L. Medintz, Medintz, Angew. Chem., Int. Ed 45 (2006) 4562-4589. DOI:10.1002/anie.200503873 |
| [6] |
K. Ema, M. Inomata, Y. Kato, H. Kunugita, M. Era, Phys. Rev. Lett. 100 (2008) 257401. DOI:10.1103/PhysRevLett.100.257401 |
| [7] |
L. Shi, V. De Paoli, N. Rosenzweig, Z. Rosenzweig, J. Am. Chem. Soc. 128 (2006) 10378-10379. DOI:10.1021/ja063509o |
| [8] |
T. Förster, Naturwissenschaften 33 (1946) 166-175. DOI:10.1007/BF00585226 |
| [9] |
D.L. Dexter, J. Chem. Phys. 21 (1953) 836-850. DOI:10.1063/1.1699044 |
| [10] |
V. Balzani, P. Ceroni, A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, Weinheim, 2014.
|
| [11] |
R.B. Sekar, A. Periasamy, J. Cell Biol. 160 (2003) 629-633. DOI:10.1083/jcb.200210140 |
| [12] |
N. Komatsu, K. Aoki, M. Yamada, et al., Mol. Biol. Cell 22 (2011) 4647-4656. DOI:10.1091/mbc.e11-01-0072 |
| [13] |
H. Sahoo, J. Photochem. Photobiol. C 12 (2011) 20-30. DOI:10.1016/j.jphotochemrev.2011.05.001 |
| [14] |
J. Ju, C. Ruan, C.W. Fuller, A.N. Glazer, R.A. Mathies, Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 4347-4351. DOI:10.1073/pnas.92.10.4347 |
| [15] |
V.V. Didenko, BioTechniques 31 (2001) 1106-1121. DOI:10.2144/01315rv02 |
| [16] |
J. Geri, J. Oakley, T. Reyes-Robles, et al., Science 367 (2020) 1091-1097. DOI:10.1126/science.aay4106 |
| [17] |
P. Werther, K. Yserentant, F. Braun, et al., bioRxiv (2020). DOI:10.1101/2020.08.07.241687v1 |
| [18] |
R.A. Marcus, J. Chem. Phys. 24 (1956) 966-978. DOI:10.1063/1.1742723 |
| [19] |
S. Dadashi-Silab, S. Doran, Y. Yagci, Chem. Rev. 116 (2016) 10212-10275. DOI:10.1021/acs.chemrev.5b00586 |
| [20] |
W. Chi, J. Chen, W. Liu, et al., J. Am. Chem. Soc. 142 (2020) 6777-6785. DOI:10.1021/jacs.0c01473 |
| [21] |
P.A. de Silva, N.H. Gunaratne, C.P. McCoy, Nature 364 (1993) 42-44. DOI:10.1038/364042a0 |
| [22] |
A.P. de Silva, T.S. Moody, G.D. Wright, Analyst 134 (2009) 2385-2393. DOI:10.1039/b912527m |
| [23] |
E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata, T. Nagano, J. Am. Chem. Soc. 127 (2005) 3684-3685. DOI:10.1021/ja042967z |
| [24] |
T. Ueno, Y. Urano, H. Kojima, T. Nagano, J. Am. Chem. Soc. 128 (2006) 10640-10641. DOI:10.1021/ja061972v |
| [25] |
F.M. Raymo, M. Tomasulo, Chem. Soc. Rev. 34 (2005) 327-336. DOI:10.1039/b400387j |
| [26] |
L. Flamigni, B. Ventura, M. Tasior, et al., Chem. Eur. J. 14 (2008) 169-183. DOI:10.1002/chem.200700866 |
| [27] |
L. Flamigni, D. Wyrostek, R. Voloshchuk, D.T. Gryko, Phys. Chem. Chem. Phys. 12 (2010) 474-483. DOI:10.1039/B916525H |
| [28] |
F. D'Souza, P.M. Smith, M.E. Zandler, et al., J. Am. Chem. Soc. 126 (2004) 7898-7907. DOI:10.1021/ja030647u |
| [29] |
V. Navarro-Pérez, A.M. Gutiérrez-Vílchez, J. Ortiz, et al., Chem. Commun. 55 (2019) 14946-14949. DOI:10.1039/C9CC07649B |
| [30] |
Y. Cai, L. Du, K. Samedov, et al., Chem. Sci. 9 (2018) 4662-4670. DOI:10.1039/C8SC01170B |
| [31] |
Q. Qi, S. Jiang, Q. Qiao, et al., Chin. Chem. Lett. 31 (2020) 2985-2987. DOI:10.1016/j.cclet.2020.05.044 |
| [32] |
P. Klan, J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice. Chichester: Wiley, 2009.
|
| [33] |
Y.J. Wang, Z. Li, J. Tong, et al., J. Mater. Chem. C 3 (2015) 3559-3568. |