 2021, Vol. 32
2021, Vol. 32 


b Department of Medical Ultrasound, Shanghai Tenth People's Hospital, Ultrasound Research and Education Institute, Tongji University School of Medicine, Tongji University Cancer Center, Shanghai 200072, China;
c Peking University Shenzhen Institute, Shenzhen 518055, China;
d State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau 999078, China;
e State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
Cancer immunotherapy eliciting the protective immune response for tumor eradication has recently received widespread attention [1]. In particular, immune checkpoint blockade (ICB) therapy has achieved promising success in clinic [2]. Up to date, several monoclonal antibody-based immune checkpoint inhibitors have been approved for clinical application of ICB therapy, including the antibodies against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (e.g., ipilimumab), programmed cell death protein 1 (PD-1) (e.g., pembrolizumab and nivolumab), and programmed death ligand 1 (PD-L1) (e.g., atezolizumab) [3-5]. Nevertheless, the therapeutic performance of ICB-based immunotherapy is restricted by several distinct challenges including the low immunogenicity of the tumor cells and the immunosuppressive tumor microenvironment (ITM) [6-11]. It is essential to give priority to develop novel strategies for improving cancer immunotherapy by promoting intratumoral infiltration of cytotoxic T lymphocytes (CTLs) and attenuating the ITM for counteract immune-tolerant tumors [12].
It has been well established that inhibition of the cyclin-dependent kinases 4 and 6 (CDK4/6) can regress tumor growth by inducing G1 cell cycle arrest through inhibiting phosphorylation of the retinoblastoma tumor suppressor [13, 14]. Recent studies further revealed that CDK4/6 inhibition can trigger anti-tumor immunity by promoting intratumoral secretion of type Ⅲ interferons (IFN Ⅲ) and impeding the proliferation of regulatory T cells (Tregs) [15]. However, CDK4/6 inhibition promotes PD-L1 expression on the surface of the tumor cell membrane by disturbing CDK4/6-mediated phosphorylation of the speckle-type POZ protein (SPOP), which impairs the immunotherapeutic performance of CDK4/6 inhibitor [16, 17]. Despite combinatory immunotherapy of CDK4/6 inhibition with PD-L1 blockade displayed improved therapeutic benefits [18], ICB therapy by antibodies severe adverse events related to the disturbed immune balance of the normal tissues due to PD-L1 expression on the surface of pancreatic islet cells, vascular endothelium, hepatocytes and epithelium [19]. To alleviate the "on-target but off-tumor" side effects of antibody-based immune checkpoint inhibitor, it is essential to exploit a tactic to deliver checkpoint inhibitors efficiently and precisely to tumor tissue [20-23].
RNA interference (RNAi) with small interfering RNA (siRNA) represents a promising strategy for ICB therapy by silencing the interested immune checkpoints at the transcription stage [22, 24-26]. In this study, we hypothesized that the therapeutic benefits of CDK4/6 inhibition could be augmented by siRNA-mediated PD-L1 knockdown. To validate our hypothesis, we herein sought to develop tumor acidity-activatable micelleplexes for tumor-specific delivery of siRNA-PD-L1 and combinatory immunotherapy with abemaciclib (Abe), a newly approved CDK4/6 inhibitor (CDK4/6i) for clinical therapy of metastatic breast cancer. The acid-activated micelleplexes composed of hydrophilic poly(ethylene glycol) (PEG) corona, a cationic intermediate layer, and a hydrophobic core were self-assemble by triblock copolymer. This micelleplexes kept stable during the blood circulation due to presence of the PEG corona, which minimizes protein absorption and reticuloendothelial system (RES) clearance [27-29]. Once arrival at the tumor site, the micelleplexes were specifically activated/dissociated in the acidic lysosome/endosome vesicles through protonation of the tertiary amine segment, for cytosol release of siRNA and subsequent silence of endogenous PD-L1 protein [30-33]. The PDPA micelleplexes with pKa value of 6.2 displayed remarkable PD-L1 knockdown efficacy. Combinatory therapy by PD-L1 blockade and CDK4/6 inhibition highly efficiently regressed tumor growth (Scheme 1).

|
Download:
|
| Scheme 1. Schematic illustration of combinatory cancer immunotherapy by PD-L1 knockdown and CDK4/6 inhibition. (a) Fabrication of acid-activatable micelleplexes for siRNA-PD-L1 delivery. The micelleplexes were loaded with siRNA-PD-L1 on the intermediate layer of the micelleplexes. The micelleplexes could be activated in the endosomal acidic environment for cytosol release of the siRNA payload; (b) Mechanism demonstration of combinatory PD-L1 knockdown and CDK4/6 inhibition for promoting cancer immunotherapy. | |
{kind=link}
To screen the acid-activatable micelleplexes for efficient siRNA delivery, three acid-responsive triblock copolymers with different p Ka values were synthesized by mPEG113-Br-initiated sequential atom transfer radical polymerization (ATRP) of glycidyl methacrylate (GA) and 2-(hexamethylamino)ethyl methacrylate (C7A), 2-(diisopropylamino)ethyl methacrylate (DPA), or 2-(dibutylamino)ethyl methacrylate) (C4A) (Fig. S1 in Supporting information) [34, 35]. The polymerization degree of the resultant triblock copolymers including poly(ethylene glycol)-block-poly(glycidyl methacrylate)-block-poly(2-(hexamethylamino)ethyl methacrylate) (PEG113-b-PGA50-b-PC7A55), poly(ethylene glycol)-block-poly(glycidyl methacrylate)-block-poly(2-(diisopropyl amino) ethyl methacrylate) (PEG113-b-PGA50-b-PDPA52), and poly(ethylene glycol)-block-poly(glycidyl methacrylate)-block-poly(2-(dibutylamino) ethyl methacrylate) (PEG113-b-PGA50-b-PC4A53) were determined by proton nuclear magnetic resonance (1H NMR) spectrum examination (Figs. S3 and S4 in Supporting information).
Afterwards, the PGA segment of PEG113-b-PGA50-b-PC7A55, PEG113-b-PGA50-b-PDPA52 and PEG113-b-PGA50-b-PC4A53 triblock copolymers were aminated with ethylene diamine (EDA) to obtain cationic triblock copolymers (Fig. S2 in Supporting information). An acid-insensitive analog of above triblock copolymers was also synthesized by PEG-PGA-Br-initiated polymerization of butyl methacrylate (BMA) and amination with EDA. The resultant PEG113-b-PEDA50-b-PBMA49 triblock copolymer was termed as PBMA (Figs. S2 and S5 in Supporting information).
The micellar nanoparticles were then prepared with above triblock copolymers by solvent precipitation method as reported previously [36]. These micellar nanoparticles displayed averaged hydrodynamic diameter around 40−60 nm determined by dynamic light scattering (DLS) analysis (Figs. 1a-d). All the micellar nanoparticles displayed positive surface charge ~18 mV due to presence of the aminated PGA interlayer in the micellar nanostructures (Figs. 1a-d). Transmission electron microscopy (TEM) examination showed spherical morphology of all the micelles nanoparticles with averaged particle size about 40–60 nm at pH 7.4, in good agreement with that examined by DLS measurement (Figs. 1e-h). The PC7A, PDPA and PC4A micelles dissociated at acidic pH to form amorphous aggregates due to protonation of the C7A, DPA and C4A tertiary amines and dissociation of the hydrophobic core (Figs. 1i-k). In contrast, PBMA displayed constant morphology and hydrodynamic diameter at both neutral pH of 7.4 and acidic pH of 5.0 since the BMA monomer is insensitive to the acidic condition (Fig. 1l). The acid-induced dissociation of these micelles at different conditions further demonstrated superior acid-sensitivity of the PC7A, PDPA and PC4A micellar nanoparticles (Fig. S6 in Supporting information). The pH titration assay showed narrow pH-transition range of the PC7A, PDPA and PC4A triblock copolymers, verifying the superior acid sensitivity of the tertiary amino groups (Fig. S7 in Supporting information). The apparent p Ka values of the PC7A, PDPA and PC4A triblock copolymers were determined as 6.8, 6.3 and 5.4, respectively.

|
Download:
|
| Fig. 1. Physiochemical characterization of the acid-responsive micelles. (a–d) DLS-determined hydrodynamic particle size distribution and zeta potential of (a) PC7A, (b) PDPA, (c) PC4A and (d) PBMA at neutral pH 7.4; (e–l) The representative TEM image of (e) PC7A, (f) PDPA, (g) PC4A at pH 7.4, (i) PC7A, (j) PDPA and (k) PC4A at pH 5.0, respectively; (h) PBMA at pH 7.4 and (l) PBMA at pH 5.0 (scale bar =50 nm); (m–p) Fluorescence spectra and fluorescent images of NR-loaded PC7A, PDPA, PC4A and PBMA micelles at different pH conditions, the insert carton images demonstrated acid-triggered dissociation of the micellar nanoparticles. | |
{kind=link}
To demonstrate acid-induced dissociation of the micelle nanoparticles, a polarity-sensitive fluorescent dye nile red (NR) was loaded into the hydrophobic core of the micellar nanoparticles [37]. NR displays strong fluorescence emission at hydrophobic environment, while quenched fluorescence signal in aqueous phase. As expected, strong fluorescence signal was observed at pH≥7.0 suspension of PC7A micellar nanoparticles, verifying stable encapsulation of NR in the hydrophobic core of the PC7A micelles. In contrast, the fluorescence emission of NR was quenched at pH≤6.8 due to acid-triggered protonation of the C7A tertiary amines and subsequent dissociation of the micellar nanoparticles (Fig. 1n). Similar phenomena were observed for PDPA and PC4A micelles at pH range of 6.2–6.4 (Fig. 1m) and 5.0–5.2 (Fig. 1o), respectively. Meanwhile, the PBMA micelles retained constant fluorescence signal throughout the pH range, verifying that PBMA nanoparticles were insensitive to buffer pH (Fig. 1p).
The serum stability of the micelles was next investigated by incubating the nanoparticles in PBS solution containing 0%, 5% or 10% (v/v) of fetal calf serum (FBS). The hydrodynamic diameter and PDI of the micellar nanoparticles changed slightly upon 24 h incubation with 10% aqueous solution of FBS, implying the satisfying colloidal stability of the micelles due to the presence of PEG corona on the surface (Fig. 2a). The micellar nanoparticles prepared from PC7A, PDPA, PC4A and PBMA triblock copolymers all displayed IC50 over 50 μg/mL in CT26 cells as examined by methyl thiazolyl tetrazolium (MTT) assay (Fig. 2b). Noticeably, PC7A micelles displayed increased cytotoxicity than that of the PC4A and PDPA micelles.

|
Download:
|
| Fig. 2. (a) Serum stability assay of the PC7A, PDPA, PC4A and PBMA micelles in serum-containing solution, the micelles were incubated at 37 ℃ for 24 h; (b) MTT examination of the micelle cytotoxicity in CT26 tumor cells; (c) Relative hemolytic activity of the PC7A, PDPA, PC4A and PBMA micelles at various pH conditions (micelle concentration of 50 μg/mL); (d) Gel retardation assay the siRNA binding ability of PC7A, PDPA, PC4A and PBMA micelles; (e) CLSM examination of intracellular distribution of the siRNA-loaded PC7A, PDPA, PC4A and PBMA micelleplexes. The nucleus were stained with Hoechst, the lysosome vesicles were labeled by Lysotracker Red, and the siRNA were labeled with FAM. Scale bar = 30 μm. | |
{kind=link}
The endosome escape ability of these micelles was investigated via hemolytic assay at various pH conditions. PMBA micellar nanoparticles displayed negligible hemoglobin release from the red blood cells (RBC) at pH 6.2 or 5.0. In contrast, PDPA micelles caused dramatic hemoglobin release at acidic pH (e.g., 6.2) mimicking the acidic environment of the early endosome vesicles (Fig. 2c). The PC7A and PC4A micelle nanoparticles displayed comparable hemolytic activity as that of the PDPA ones at acidic pH below the p Ka values of the copolymers (i.e., 6.8 for PC7A and 5.0 for PC4A, respectively). The hemolytic ability of the PDPA, PC7A and PC4A micelles could be most likely attributed to the "proton sponge" effect of the protonated PC7A, PDPA, PC4A tertiary amines to destabilize the RBC membrane.
Gel shift assay was employed to evaluate the siRNA binding affinity of the micellar nanoparticles. A positive correlation was demonstrated between the micelle to siRNA weight ratio and their siRNA binding ability, and all the micelles showed excellent ability for compressing siRNA into the micelles at the copolymers to siRNA weight ratio of 8.0 (Fig. 2d). The siRNA complexation ability of the cationic micelles could be attributed to the aminolated PGA interlayer.
Given the satisfying siRNA binding ability of the cationic micelle nanoparticles, we next sought to investigate the ability of micelleplexes for intracellular siRNA delivery in vitro. All the micelleplexes loaded with FAM-labeled siRNA (siRNA-FAM) was internalized by the tumor cells at an incubation time-dependent manner, verifying the ability of the micelleplexes for efficient siRNA delivery (Fig. S8 in Supporting information).
The intracellular distribution of the siRNA-loaded micelleplexes were examined via confocal laser scanning microscope (CLSM) measurement. The micelleplexes were constructed with fluorescein-modified siRNA-NC (siRNA-FAM). In the PC7A and PDPA micelleplexes-incubated tumor cells, the green fluorescence of siRNA-FAM was not completely co-localized with lysosome vesicles and partially throughout the cytosol, indicating the siRNA escaped from endosomes successfully due to the rosy membrane instability of these triple-layered micelleplexes at acid pH mimicking the early endosome environment.
On the contrary, slight green spots of siRNA-FAM were observed in the cytosol of PC4A micelle-treated tumor cells, which can be mostly attributed to the membrane lysis ability of the micelleplex occurred at late endosome environment. The PMBA group displayed negligible green fluorescence, implying minimal cytosol release of siRNA payload since PBMA micellar nanoparticles showed low erythrocyte lysis property at varied pH conditions (Fig. 2e). These results verified the potential of the PC7A and PDPA micelleplexes for intracellular siRNA delivery and cytosol release inside the tumor cells via protonation-induced endosome escape.
To investigate the optimized micellar formulation for silencing the targeted genes, we measured the siRNA transfection capability of the micelleplexes in CT26 colorectal tumor cells with stabile green fluorescence protein (GFP) expression (CT26-GFP). Fig. 3a displayed that the siRNA transfection efficacy increased gradually at a function of micelle-to-siRNA weight ratio. For instance, about 60% and 70% of GFP expression were silenced in the PDPA and PC7A micelleplex groups (w/w=16). The PDPA and PC7A micelleplex knocked down about 70% of GFP protein expression at micelle-to-siRNA weight ratio of 32. It was worth noting that despite the cationic micelles complexed the siRNA payload at a low micelle-to-siRNA weight ratio of 6.0 (Fig. 2d), the transfection data displayed that higher micelle-to-siRNA weight ratio (e.g., > 16) is preferred for siRNA transfection in vitro. This phenomenon suggested a crucial role of free cationic micelle for endosome escape of the nucleic acid payloads as reported previously [38, 39].

|
Download:
|
| Fig. 3. Gene knockdown efficacy of the micelleplexes in CT26-GFP tumor cells in vitro. (a) Influence of micelle to siRNA weight ratio (at a siRNA concentration of 75 nmol/L), and (b) siRNA-GFP concentration (at a micelle-to-siRNA weight ratio 32) on the GFP knockdown ability of the PC7A, PDPA, PC4A and PBMA micelleplex; (c) Influences of Baf-A1 incubation on the GFP knockdown efficacy of the siRNA-GFP-loaded micelleplexes at a siRNA concentration of 75 nmol/L and micelle to siRNA weight ratio of 32; (d) CLSM examination of micelleplex-mediated GFP knockdown in CT26-GFP cells in vitro (Scale bar = 100 μm); (e) Western blot assay of PDPA-siRNA-PD-L1 micelleplex-mediated PD-L1 knockdown in CT26 tumor cells in vitro; (f) Quantification of PD-L1 knockdown efficacy of the PDPA/siRNA-PD-L1 micelleplexes in vitro (**** P < 0.0001). | |
{kind=link}
Furthermore, both PDPA and PC7A micelleplexes displayed siRNA dose-dependent GFP knockdown efficacy at a constant micelle-to-siRNA weight ratio of 32 (Fig. 3b). The results verified that over 50% of GFP expression were silenced, especially PDPA and PC7A micelleplex displayed stronger gene knockdown capability than PC4A micelleplex at the siRNA dose of 0.5 μg/mL. PDPA and PC7A micelleplex dramatically silenced about 70% of GFP expression when the siRNA dose increased up to 1.0 μg/mL (Fig. 3b). In contrast, PMBA micelleplex displayed negligible GFP knockdown efficacy at various conditions (Figs. 3a and b). The results could be most likely attributed to protonation of the pH-response PC7A, PDPA and PC4A core caused the disruption of lysosomes, the released siRNA subsequently silenced GFP knockdown in vitro. In contrast, the protonation absent PBMA had negligible affection on GFP expression. With respect to the different efficient of gene knockdown, especially PC4A micelleplex showed suboptimal capability of gene knockdown, mostly attribute to different pKa value of their triple copolymer, determining their membrane disruption capability at various acid condition mimicking the early endosome or late endosome/lysosome.
To further demonstrate the influence of endosomal acidification on the gene knockdown efficacy of the micelleplexes, CT26 cells were pre-incubated with bafilomycin-A1 (Baf-A1), a potent vacuolar-type H+ ATPase (V-ATPse) proton pump inhibitor [40]. Both siRNA-GFP micelleplexes silenced < 20% of GFP expression with the pre-treatment of Baf-A1. In contrast, the GFP expression were obviously detected when absent pre-incubation with Baf-A1. It mostly explained by the dissociation of micelleplexes via the protonation of PC7A, PDPA and PC4A core induced by acidic condition were inhibited through Baf-A1, and therefore restrained the siRNA-GFP escape from the endosome (Fig. 3c). These results were further verified by CLSM examination, in which the siRNA-GFP or siRNA-NC were loaded to the different micelles. For instance, the green fluorescence was almost invisible at the PDPA-siRNA-GFP and PC7A-siRNA-GFP groups, the slightly green fluorescence was observed at the PC4A-siRNA-GFP group without Baf-A1-treated. In contrast, the green fluorescence was almost no distinction upon treated with Baf-A1. In regard to the PBMA micelleplexes, GFP expression were observed distinctly of neither loaded with siRNA-NC nor the siRNA-GFP and even pre-treated with Baf-A1 (Fig. 3d).
Although PDPA and PC7A micelleplexes displayed comparable GFP knockdown efficacy (Fig. 3c), the PDPA micellar nanoparticles showed lower cytotoxicity than the PC7A micelleplexes (Fig. 2b). We thus selected PDPA micelleplexes prepared at a micelle-to-siRNA weight ratio of 32 for PD-L1 knockdown in vitro and in vivo. Western blotting assay displayed that the PDPA-siRNA-PD-L1 micelleplexes efficiently downregulated PD-L1 expression in the CT26 tumor cells in siRNA dose-dependent manner (Fig. 3e). Over 50% of the endogenous PD-L1 expression was silenced with PDPA-siRNA-PD-L1 micelleplexes at a siRNA concentration of 100 nmol/L (Fig. 3f). This confirmed the ability of the acid-activatable PDPA micelleplexes for PD-L1 knockdown in vitro.
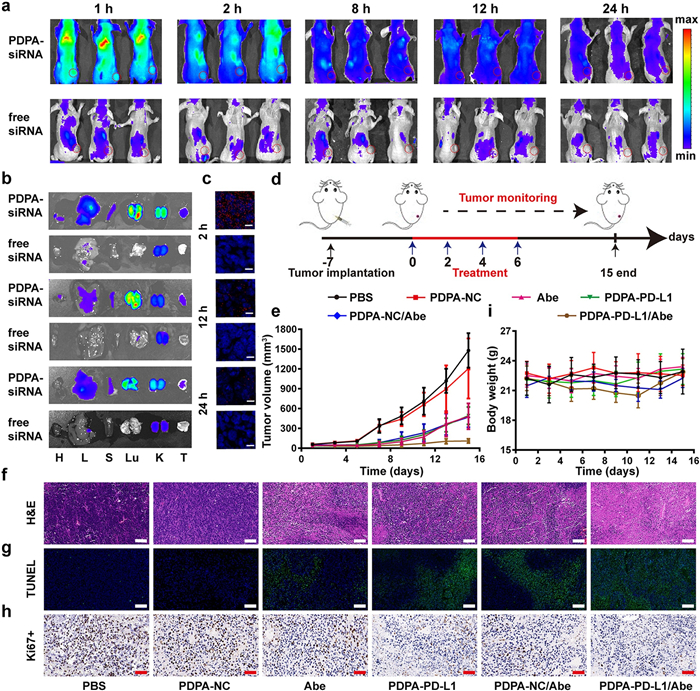
The systemic biodistribution of the siRNA-loaded PDPA micelleplexes was investigated in CT26 tumor-bearing Balb/c mouse model in vivo. The PDPA micelleplex was prepared with the Cy5.5-labeled siRNA-NC (termed as PDPA-siRNA), and systematically administrated via intravenous (i.v.) injection at the dose of 2.0 mg/kg of siRNA when the tumor volume reached ~200 mm3. The biodistribution of the siRNA-loaded micelleplexes was detected by fluorescence imaging in vivo at predetermined time points postinjection. In compared with free siRNA, the fluorescence signal of the PDPA-siRNA was detected throughout the experimental period, most likely due to the micelleplex can protect siRNA from rapid blood clearance during circulation. Meanwhile, the fluorescence signal at tumor tissues was evident even post-injected around 24 h, indicating the strong tumor accumulation via enhanced permeability and retention (EPR) effect (Fig. 4a). The tumor tissues were harvested at the 2 h, 12 h or 24 h post-injection and freeze sectioned for CLSM examination of the intratumoral distribution of the micelleplex ex-vivo. Fluorescence imaging of the tumor organs ex-vivo further validated increased intratumoral siRNA distribution of the PDPA-siRNA group than that of the free siRNA control (Fig. 4b). CLSM examination of the tumor sections further validated increased intratumoral siRNA distribution that of the free siRNA, in consistence with the data accumulation by fluorescence imaging ex-vivo (Fig. 4c).

|
Download:
|
| Fig. 4. Biodistribution and antitumor efficacy of the micelleplex. (a) Fluorescence examination about biodistribution of micelleplex loaded with siRNA-NC-Cy5.5 and free siRNA-NC-Cy5.5 in CT26 tumor-bearing mice; (b) Ex-vivo fluorescence imaging of the major tissues at 2 h, 12 h and 24 h (H: heart; L: liver; S: spleen; Lu: lung; K: kidney; T: tumor); (c) Immunofluorescence staining of intratumoral siRNA-NC-Cy5.5 distribution examined at 2 h, 12 h and 24 h post tail-vein injection, respectively; (d) Therapeutic schedule of the combinatory immunotherapy of CT26 tumor in vivo; (e) Tumor growth profile of the CT26 tumor-bearing mice with the indicated treatments (n=6, mean±SD); (f) H & E, (g) TUNEL staining, and (h) immunohistochemical examination of Ki-67+ expression of the tumor sections at the end of antitumor studies (scale bar = 100 μm); (i) Body weight change of the CT26 tumor-bearing mice recorded during the antitumor studies (n = 6, mean±SD). | |
{kind=link}
CDK4/6i has been demonstrated to arrest the tumor cells in the G1 phase and further elicit anti-tumor immunity. Abe (CDK4/6i), has been approved by Food and Drug Administration for tumor therapy, showed obvious cell cytotoxicity in vitro (Fig. S9 in Supporting information). Nonetheless, CDK4/6 inhibition also induced PD-L1 up-regulation via phosphorylation of the speckle-type POZ protein [41]. Thus, siRNA-PD-L1-loaded PDPA micelleplexes (termed as PDPA-PD-L1) were selected to enhance the therapeutic efficacy of CDK4/6i Abe by silencing both endogenous and inducible PD-L1 expression on the surface of the tumor cell membrane. The combination of PD-L1 knockdown and CDK4/6 inhibition can not only suppress the growth of tumor but also activate the anti-tumor immunity via exerting the synergistic effect, and meanwhile compensating the negative feedback of Abe to realized more excellent anti-tumor efficacy. The antitumor efficacy of micelleplex-mediated PD-L1 silence and Abe-based CDK4/6 blockade was evaluated in xenograft CT26 tumor-bearing Balb/c mouse model. The mice were randomly divided into 6 groups when the tumor volume reached 100 mm3. The tumor-bearing mice were intravenously (i.v.) administrated with the siRNA-NC-loaded PDPA micelleplex (termed as PDPA-NC), PDPA-PD-L1 at the identical siRNA dose of 0.6 mg/kg, and Abe was intraperitoneal injected at a dose of 50 mg/kg, PDPA-NC/Abe and PDPA-PD-L1/Abe, respectively. The treatment was repeated for 4 times at every 2 days, the tumor volume and body weight were monitored continuously during the experimental period (Fig. 4d).
Monotherapy by Abe or siRNA-PD-L1 alone moderately inhibited tumor growth. In contrast, the combination of Abe with PDPA-PD-L1 knockdown dramatically regressed tumor growth (Fig. 4e). Hematoxylin-eosin (H & E) staining and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) analysis of the CT26 tumor sections verified that the combination of Abe and PDPA-PD-L1 significantly induced apoptosis and necrosis of the tumor cells (Figs. 4f and g). Combinatory therapy by PDPA-PD-L1 and Abe showed the lowest Ki-67+ ratio of the tumor sections compared to that of the control groups (Fig. 4h). There was no obvious body weight loss (Fig. 4i). H & E staining of the major organs (heart, liver, spleen, lung and kidney) revealed inappreciable histological damage, implying desirable biosafety and satisfied biocompatibility of the micelleplex (Fig. S10 in Supporting information). All these results indicated that combination of PD-L1 knockdown and CDK4/6 inhibition in the tumor cells not only could disrupt tumor cell directly but also trigger anti-tumor immunity, resulting in obvious improvement of the antitumor efficacy.
To elucidate the mechanism underlying the remarkable anti-tumor efficacy of the combination therapy, we firstly examined PD-L1 expression by immunohistochemical and immunofluorescence staining of the tumor sections ex-vivo. Figs. 5a and b revealed that the Abe treatment upregulated PD-L1 expression undesirably. Interestingly, PD-L1 knockdown with the micelleplexes perfectly offset Abe-induced PD-L1 expression, indicating the admirably synergistic therapeutic efficacy for tumor.

|
Download:
|
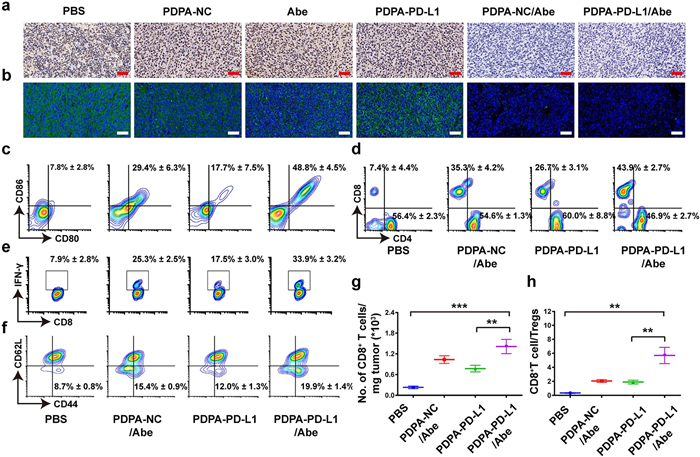
| Fig. 5. Immune mechanism analysis of M-siRNA-PD-L1/Abe combination therapy. (a) Immunohistochemical and (b) immunofluorescence staining of intratumoral PD-L1 expression at the end of antitumor study (Blue: Hoechst; Green: PD-L1; Scale bar =100 μm); (c–f) Flow cytometry examination of (c) DC maturation (gated by CD11+CD80+CD86+) in the tumor-draining LNs, (d) intratumoral infiltration of CD8+ and CD4+ T cells (gated by CD3), (e) IFN-γ+CD8+ T cells and (f) frequency of CD8+CD44+CD62L− memory T lymphocytes in the spleen of CT26 tumor-bearing BALB/c mice with various treatments; (g) Tumor mass-normalized tumor-infiltrating CD8+ T cells; (h) The ratio of tumor-infiltrating CD8+ T cell to Tregs (n=3, mean±SD; **P < 0.01; ***P < 0.001). | |
{kind=link}
To further explore the mechanism of the antitumor effect of combinatory therapy by PD-L1 knockdown and CD4/6 inhibition, we next evaluated DC maturation in the tumor-draining lymph nodes by flow cytometry analysis. The PDPA-NC/Abe and PDPA-PD-L1 groups displayed 3.7- and 2.2-fold higher DC maturation ratio (CD11c+CD86+CD80+) than that of the PBS groups respectively, demonstrating the Abe and PDPA-PD-L1 mediated PD-L1blockade both can promote the maturation of DCs. Correspondingly, the combination group of PDPA-PD-L1/Abe was significantly increased the frequency of matured DCs, which showed 6.2-fold higher than PBS group (Fig. 5c).
The matured DCs can present tumor antigens to activate the tumor-specific CD8+ T cells for tumor regression [42, 43]. The PDPA-PD-L1/Abe group displayed 5.9-fold higher CD8+ to CD4+ T cells ratio as compared to that of the PBS group (Fig. 5d). The tumor mass-normalized tumor-infiltrating CD8+ T cells also showed the similar results, indicating that the combinatory therapy by PDPA-PD-L1/Abe remarkably promoted the intratumoral infiltration of CD8+ T cells (Fig. 5g). Notably, the frequency of IFN-γ+ CD8+ T cells of the PDPA-PD-L1/Abe group distinctly increased to 33.85%, which was 6.8-fold higher than that of the PBS group. Furthermore, the frequency of the IFN-γ+CD8+ T lymphocytes of the PDPA-PD-L1 and PDPA-NC/Abe group increased by 3.5- and 5.1-fold compared to that of the PBS group (Fig. 5e).
The activation of immune memory effect was crucial for durable tumor regression and metastasis prevention, we thus sought to investigate the frequency of the effector memory T cells (TEM, CD3+CD8+CD44+CD62L−) in the spleen. The ratio of CD8+ TEM in the PDPA-PD-L1 and PDPA-NC/Abe groups both increased obviously. For instance, PDPA-PD-L1/Abe group showed 2.3-fold higher CD8+ TEM compared to that of the PBS group, validating the potential of combinatory therapy by PDPA-PD-L1/Abe to prevent tumor metastasis and reoccurrence (Fig. 5f).
The regulatory T cells (Tregs, CD4+Foxp3+CD25+ T lymphocytes) is considered relate to the immunosuppressive microenvironment due to suppress the activation of CD8+ T cells [44]. Hence, the intratumoral infiltrating Tregs were investigated upon different treatment. Fortunately, the PDPA-NC/Abe or PDPA-PD-L1 group both can decrease the proportion of Treg in various degree (Fig. S11 in Supporting information). Furthermore, combinatory therapy by PDPA-PD-L1/Abe group reduced Tregs ratio distinctly by 2.2-fold lower than that PBS group (Fig. S10 in Supporting information). The CD8+T cell-to-Tregs ratio in PDPA-PD-L1/Abe group was 16.7- and 2.7-fold higher than control group and PDPA-NC/Abe group respectively, indicating the combination of PD-L1 blockade and Abe dramatically relieve the immunosuppress microenvironment (Fig. 5h).
In summary, we herein reported a series of acid-activatable micelleplexes for tumor-specific siRNA delivery and PD-L1 knockdown in the tumor cells. The micelleplexes were further employed for combinatory therapy with CDK4/6i for improving cancer immunotherapy. The PDPA micelleplexes with rationally optimized p Ka value of 6.3 displayed satisfying PD-L1 knockdown efficacy and good biocompatibility. In combination with CDK4/6 inhibition with Abe, the PDPA micelleplexes dramatically suppressed inducible PD-L1 expression in the tumor cells, thus promoted intratumoral infiltration of CTLs, activated immune memory T lymphocytes, and relived the ITM for improved tumor regression. This study suggested the potent potential of combinatory therapy by PD-L1 knockdown and CDK4/6 blockade for promoting cancer immunotherapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 51873228 and 31671024), Basic Research Program of Shenzhen (No. JCYJ20180227175420974), Science and Technology Development Fund, Macau SAR (No. 083/2017/A2), and Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. All animal procedures were carried out under the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.12.009.
| [1] |
R.S. Riley, C.H. June, R. Langer, et al., Nat. Rev. Drug Discov. 18 (2019) 175-196. |
| [2] |
M.C. Andrews, J.A. Wargo, Cell 171 (2017) 740-742. DOI:10.1016/j.cell.2017.10.027 |
| [3] |
J.D. Wolchok, H. Kluger, M.K. Callahan, et al., N. Engl. J. Med. 369 (2013) 122-133. DOI:10.1056/NEJMoa1302369 |
| [4] |
P.C. Tumeh, C.L. Harview, J.H. Yearley, et al., Nature 515 (2014) 568-571. DOI:10.1038/nature13954 |
| [5] |
R.S. Herbst, J.C. Soria, M. Kowanetz, et al., Nature 515 (2014) 563-567. DOI:10.1038/nature14011 |
| [6] |
T.F. Gajewski, H. Schreiber, Y.X. Fu, Nat. Immunol. 14 (2013) 1014-1022. |
| [7] |
S.J. Turley, V. Cremasco, J.L. Astarita, Nat. Rev. Immunol. 15 (2015) 669-682. |
| [8] |
P.S. Hegde, V. Karanikas, S. Evers, Clin. Cancer Res. 22 (2016) 1865-1874. DOI:10.1158/1078-0432.CCR-15-1507 |
| [9] |
A. Gao, Hu Xl., M. Saeed, et al., Acta Pharmacol. Sin. 40 (2019) 1129-1137. |
| [10] |
A. Gao, B. Chen, J. Gao, et al., Nano Lett. 20 (2020) 353-362. |
| [11] |
B. Feng, B. Hou, Z. Xu, et al., Adv. Mater. (2019) 1902960. |
| [12] |
B. Feng, Z. Niu, B. Hou, et al., Adv. Funct. Mater. (2019) 1906605. |
| [13] |
J.M.R. Charles, J. Sherr, Genes Dev. 13 (1999) 1501-1512. |
| [14] |
Y.J. Choi, X. Li, P. Hydbring, et al., Cancer Cell 22 (2012) 438-451. |
| [15] |
S. Goel, M.J. DeCristo, A.C. Watt, et al., Nature. 548 (2017) 471-475. |
| [16] |
J. Zhang, X. Bu, H. Wang, et al., Nature. 553 (2017) 91-95. |
| [17] |
S.O. Lim, C.W. Li, W. Xia, et al., Cancer Cell 30 (2016) 925-939. |
| [18] |
S.C. Wei, J.H. Levine, A.P. Cogdill, et al., Cell 170 (2017) 1120-1133. |
| [19] |
A.H. Sharpe, E.J. Wherry, R. Ahmed, et al., Nat. Immunol. 8 (2007) 239-245. |
| [20] |
Y. Mi, C.C. Smith, F. Yang, et al., Adv. Mater. (2018) 1706098. |
| [21] |
Q. Zhu, M. Saeed, R. Song, et al., Chin. Chem. Lett. 31 (2020) 1051-1059. |
| [22] |
W. Wang, M. Saeed, Y. Zhou, et al., J. Gene Med (2019) e3092. |
| [23] |
J. Ma, J. Zhang, L. Chi, et al., Chin. Chem. Lett. 31 (2020) 1427-1431. |
| [24] |
A. Fougerolles, H.P. Vornlocher, J. Maraganore, et al., Nat. Rev. Drug Discov. 6 (2007) 443-453. |
| [25] |
J.J.R. Gregory, J. Hannon, Nature 431 (2004) 371-378. |
| [26] |
J. Gao, W.Q. Wang, Q. Pei, et al., Acta Pharmacol. Sin. 7 (2020) 986-994. |
| [27] |
X. Duan, C. Chan, W. Lin, Angew. Chem. Int. Ed. 58 (2019) 670-680. |
| [28] |
W. Yu, M. Shevtsov, X. Chen, et al., Chin. Chem. Lett. 31 (2020) 1366-1374. |
| [29] |
Z. Shi, Q. Li, L. Mei, Chin. Chem. Lett. 31 (2020) 1345-1356. |
| [30] |
H. Yu, C. Guo, B. Feng, et al., Theranostics 6 (2016) 14-27. |
| [31] |
H.J. Yu, Y.G. Wang, X.N. Huang, et al., ACS Nano 5 (2011) 9246-9255. |
| [32] |
J. Gao, W.Q. Wang, H.J. Yu, Acta Polym. Sin. 50 (2019) 1156-1166. |
| [33] |
L.Q. Maksym, V. Yezhelyev, R.M. Regan, et al., J. Am. Chem. Soc. 130 (2008) 9006-9012. |
| [34] |
H. Yu, Y. Zou, Y. Wang, et al., ACS Nano 5 (2011) 9246-9255. |
| [35] |
H. Yu, Z. Xu, D. Wang, et al., Polymer Chem. 4 (2013) 5052-5055. |
| [36] |
P. Yu, H. Yu, C. Guo, et al., Acta Biomater. 14 (2015) 115-124. |
| [37] |
H. Yu, Z. Xu, D. Wang, et al., Polym. Chem. 4 (2013) 5052-5055. |
| [38] |
Y. Hu, Z. He, Y. Hao, et al., ACS Nano 13 (2019) 10161-10178. |
| [39] |
S. Boeckle, K. Gersdorff, S. Piepen, et al., J. Gene Med. 6 (2004) 1102-1111. |
| [40] |
L.S. Johnson, K.W. Duun, B. Pytowski, et al., J. Mol. Cell. 4 (1993) 1251-1266. |
| [41] |
A. Patnaik, L.S. Rosen, S.M. Tolaney, et al., Cancer Discov. 6 (2016) 740-753. |
| [42] |
D.S. Chen, I. Mellman, Immunity 39 (2013) 1-10. |
| [43] |
M. Saeed, J. Gao, Y. Shi, et al., Theranostics 9 (2019) 7981-8000. |
| [44] |
S. Sakaguchi, M. Miyara, C.M. Costantino, et al., Nat. Rev. Immunol. 10 (2010) 490-500. |