 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Pesticide & Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, China;
c School of Pharmacy, Tongji Medical College of Huazhong University of Science & Technology, Wuhan 430030, China;
d Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, Lexington, KY 40536, United States
4-Hydroxyphenylpyruvate dioxygenase (HPPD) is a member of Fe(Ⅱ)-dependent α-keto dioxygenase that catalyzes the conversion of 4-hydroxyphenylpyruvic acid (HPPA) to homogentisic acid (HGA). HPPD can also catalyze the conversion of other α-keto acid, such as phenylpyruvate and isocaproate to 2-hydroxyphenylacetate and β-hydroxy β-methylbutyrate, respectively [1, 2]. Featured in aerobic forms of life, HPPD participates in the tyrosine catabolism, which is necessary for the photosynthesis process. As an effective target for herbicides [3-5] and promising target for the treatment of type Ⅰ tyrosinemia and alkaptonuria [6], HPPD attracts much research interests.
Numerous studies were performed to unveil the catalytic mechanism of HPPD [7-11] as well as other α-keto acid dioxygenases. Different to mechanism of majority α-keto dioxygenases that have three substrates (Scheme S1 in Supporting information), HPPD has only two substrates, i.e., HPPA and dioxygen, to accomplish all the chemistry that common to this family. The commonly accepted mechanism is that the catalysis is initiated by the bidentate binding of HPPA with the ferrous center in the HPPD active site, followed by the oxygen addition, keto acid decarboxylation, phenyl ring hydroxylation, side chain migration, and dissociation of the product HGA (Scheme 1). The reported catalytic constants (kcat) for the conversion of HPPA catalyzed by wild-type HPPD range from 1.1 s−1 to 7.8 s−1 [10, 12-14]. According to the transition state theory, the activation free energy for this reaction is about 16−17 kcal/mol. These studies also showed that among those steps the phenyl ring hydroxylation step should be rate-determining [9, 11, 15].

|
Download:
|
| Scheme 1. The common catalytic mechanism for the conversion of HPPA catalyzed by HPPD. | |
Similar to other α-keto dioxygenases, the HPPD shows ordered substrate/inhibitor binding. The association of HPPA or HPPD inhibitor is always prior to the binding of the dioxygen. It was observed that the association of the HPPA greatly induced the reactivity toward dioxygen [16]. Most HPPD inhibitors mimic substrate HPPA. However, experimental studies indicated that the HPPD-inhibitor-Fe(Ⅱ) complexes performed poorly in associating the molecule dioxygen [17].
Recently, we reported the crystal structure of Arabidopsis thaliana HPPD (AtHPPD) complexed with the substrate HPPA [12] and a cobalt ion (Fig. 1). In this complex, the pyruvate moiety of HPPA in a bidentate chelation with the cobalt ion leads to an octahedral coordination geometry involving the facial triad. The phenolic hydroxyl forms a hydrogen bond with the side chain of Asn423. The benzene ring of HPPA shapes a T-π interaction with Phe381. Compared to the other reported AtHPPD structures (PDB ID: 1SQD, 1SP9 and 1TG5) [18, 19], the HPPD-HPPA-Co(Ⅱ) structure mainly shows the following differences: 1) a conformational alteration for residue Phe428 on the C-terminal α-helix, 2) β-sheet fragment (Phe250-Phe253) rotates ~ 30° transforming into a loop, and 3) a conformation change for Gln293 upon the binding of HPPA. These differences reflect the flexibility of the HPPD binding pocket that can accommodate more substrate conformations, and the potential role of Gln293 in the connection of H-bond network of Ser267-Asn282-Gln307.

|
Download:
|
| Fig. 1. (A) Full view and (B) close view of the binding of HPPA at the catalytic site of the AtHPPD-HPPA crystal complex. Values are in the unit of angstrom. | |
In our previous works the bound HPPA was proven to be in keto form by hybrid quantum mechanics/molecular mechanics (QM/MM) calculations. These calculations also showed that structure of the restored HPPD-HPPA-Fe(Ⅱ) complex resembled the crystal complex structure with RMSD value 0.073 Å for the key geometrical parameters in the metal centers [ 12]. It was thought that the latter mimicked the pre-reactive HPPD-HPPA complex. However, the HPPA binding mode in this complex differs significantly to the proposed ones [20-22], in which the keto and carboxylic groups of HPPA lie respectively trans to the residue Glu394 and His226, and the phenyl ring flipps away from the residue Glu394. These differences prompted us to re-investigate the HPPD-HPPA-Co(Ⅱ) crystal complex in order to find out the nature of this structure.
In this study, the cobalt ion in the crystal structure is replaced with a ferrous ion to construct a AtHPPD-HPPA-Fe(Ⅱ) complex. Since there are unpaired electrons in the catalytic site of the restored complex, it is necessary to determine the spin state for this complex. Previous studies showed that different density functionals provide qualitatively different predictions [23] on the relative stability for the intermediate species in different spin states. The benchmark CCSD(T) calculations supported the B3LYP energetic [23].
Collected in Table 1 are the selected geometrical parameters, atomic charges and spin densities for selected atoms, and the relative Gibbs free energy for the HPPD-HPPA-O2 complex with different spin multiplicity. Part parameters are also shown in Fig. S1 (Supporting information) for comparison. In spin states such as the singlet, triplet and quintet state, the dioxygen is attached to the ferrous ion with interatomic Fe-O distances of 1.7–2.0 Å. However, in septet state the dioxygen is separated from the ferrous center with interatomic Fe-O distance of 3.6 Å. The optimized Fe-O distances in triplet, quintet and septet state are consistent with the results obtained in previous studies on α-ketoglutarate dependent oxygenase [24, 25]. According to the relative energies for the geometries obtained in different spin states (Table 1), the quintet, triplet, open-shell singlet, and singlet states lie about 0.5, 11.8, 24.1 and 28.4 kcal/mol above the ground septet state, which are thought to be a consequence of tighter binding of O2 to Fe [23]. Population analysis on septet state shows that there are respectively 4 and 2 unpaired alpha-electrons on the ferrous ion and the dioxygen, implying a strong Pauli repulsive interaction between them. The repulsive force should also resist the approaching of the dioxygen to the nearby HPPA α-KG group. Hence, the septet state should not be favorable for the upcoming dioxygen addition reaction. In quintet state, the electrons from the dioxygen are partially counter balanced by that of the iron leading to a net spin density of 0.55, consistent with the predictions of previous studies [23]. The quintet state is only ~0.5 kcal/mol less stable than the septet, and is by far more stable than the other three states by ~11−23 kcal/mol. Population analysis also shows that in quintet state the bound dioxygen is negatively charged and is in doublet spin state, which is thought to be a reactive species toward the α-KG group. Above discussions suggest that the quintet should be the most favorable spin state for the dioxygen addition reaction. This conclusion is consistent with the experimental observations [26, 27] and previous theoretical studies on the O2-activation steps of α-KG dioxygenases [9, 23], which are also most likely to be run on the quintet potential energy surface (PES).
|
|
Table 1 Key interatomic distances, atomic charges, spin densities and relative QM/MM Gibbs free energy of the ternary HPPD-HPPA-O2 complex calculated at different spin states.a |

{kind=link}
{kind=link}
Analysis mentioned above suggests that quintet state is most favorable for the reaction of bound dioxygen with HPPA. According to the accepted mechanism of HPPD catalysis described in Scheme 1, the negatively charged oxygen atom from the dioxygen, namely Oa, will attack the carbonyl carbon C2' of the α-KG group, affording an oxidized intermediate, which should be a local minimum of the potential energy surface (PES) along the reaction coordinate. During these calculations the RC2'-C3' – ROa-C2' is set as the reaction coordinate. However, the QM/MM minimum energy path scan on the PES fails to locate this local minimum. In fact, these calculations show that with the approaching of the Oa to the C2' atom (Fig. 2) the total energy of the complex structure keeps rising, demonstrating that the nucleophilic attack of the bound dioxygen is hold back by certain factors. Thus, in this ternary complex structure the bound HPPA behaves like an HPPD inhibitor by resisting the association of dioxygen.

|
Download:
|
| Fig. 2. Relative QM/MM energy profile for the attack of the bound dioxygen on the carbonyl carbon of HPPA in the ternary HPPD-HPPA-O2 complex in quintet state. The relative energy is determined at the B3LYP/6-311++G(2d, 2p): AMBER level. | |
{kind=link}
In order to locate the possible factors that hinder the approaching of the dioxygen to the α-KG group of HPPA, we perform reaction coordinate calculations on a QM-cluster constructed from the crystal complex. Surprisingly, calculations on this QM-cluster show that the dioxygen associates successfully with the HPPA (Figs. S2 and S3 in Supporting information). This suggests that the protein environment surrounding the catalytic triad should play important role in affecting the interaction of dioxygen with HPPA. By comparing the pre-reactive structure of the QM-cluster with the QM region of the HPPD-HPPA-O2 complex, it is found that the orientation of the phenyl ring profoundly affects the electrophilic attacking of the bound dioxygen. NCIPLOTs analysis (Fig. 3B) on one snapshot from the QM/MM PES scan shows that there is a significant repulsive interaction (brown region) between the incoming dioxygen and the phenyl carbon atoms, and a weak attractive interaction (blue region) between the dioxygen and the benzene hydrogen. Obviously, both interactions should hold back the nucleophilic attacking of the bound dioxygen on the α-KG group. On the other hand, the orientation of the phenyl ring is fixed by sidechains of the residue Gln379, Phe381, and Asn423 and buried water (Fig. 3A). Among these residues, the Phe381 should play a key role in restraining the rotation of the phenyl ring. B-factor analysis on the AtHPPD-HPPA crystal structure shows low temperature values for these residues (Fig. S8 in Supporting information). Hence, these aromatic side chains are not likely to rotate upon thermal fluctuations.

|
Download:
|
| Fig. 3. (A) Residues around the phenyl ring of HPPA in the optimized structure of ternary HPPD-HPPA-O2 complex. The value is in the unit of angstrom. (B) NCIPLOTs of non-bonded interactions among the attacking dioxygen, 4-hydroxyl group, and pyruvate group. The green area illustrates attractive interaction and the brown area represents repulsive interaction. The color scale is -0.04 < ρ < 0.04 a.u. | |
{kind=link}
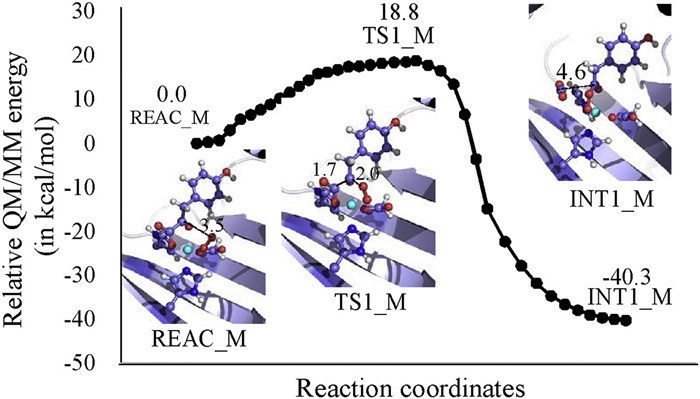
To verify the assumption mentioned above, QM/MM geometrical optimization is performed on a F381A mutant, followed by PES scan along the reaction coordinate RC2'-C3' – ROa-C2' (Fig. 4). As can be seen from this figure, in F381A mutant the Oa from the bound dioxygen gradually approaches C2' of the α-KG group and successfully covalently bond with the latter. During the forming of the Oa-C2' bond, the C3' atom departs from the C2' atom, leading to the release of a carbon dioxide. Meanwhile, the HPPA phenyl ring rotates ~40°, which is three times it does in the wild-type enzyme (Fig. S4 in Supporting information). Noteworthily, the mechanism described here is different to the one obtained from the QM-cluster calculations (Fig. S2 in Supporting information), in which addition of the dioxygen and departure of the carbon dioxide are taken place in a stepwise manner. This suggests that the position of the phenyl ring may also affect the shape of potential energy profile for the reaction of the bound dioxygen with the α-KG group. Free energy barrier and free energy change for this step are respectively calculated to be 18.8 and -40.3 kcal/mol, demonstrating that the dioxygen will readily be associated with the substrate HPPA.

|
Download:
|
| Fig. 4. Relative energy profile and geometries of the stationary points for the dioxygen addition step in F398A mutant. The total charge and multiplicity for the QM region are set to be 0 and quintet, respectively. The relative energy is determined at the B3LYP/6-311++G(2d, 2p): AMBER level. | |
{kind=link}
It is necessary to address the role of the residue Phe381 during the catalysis in order to characterize the HPPD-HPPA complex observed in the crystal. Kinetic experiments [12] showed that the Km for F381A mutant was 9.1 μmol/L, whereas the one for the wild-type HPPD was 1.9 μmol/L. The F381A mutant had similar kcat (0.92 s−1) to that of the wild-type (1.08 s−1). These data suggest that Phe381 may not directly participate the catalytic reaction. Hence, HPPA observed in the crystal structure should be a temporary intermediate during the substate transportations, instead of the previously assumed pre-reactive configuration. As HPPA is transported into pre-reactive configuration, it will be readily converted to HGA and released to the environment, leaving an empty catalytic pocket. As evidenced by the crystal structure reporting the bound HPPA (Fig. S11 in Supporting information), three out of four subunits are vacant, implying that most conversion reactions in these places have completed. The present study demonstrates that the HPPA binding mode observed in the crystal structure is not the reactive mode. Despite various binding modes advanced so far, more proofs are still needed to validate them. So far, it is still an open question for the determine the reactive binding mode of HPPA.
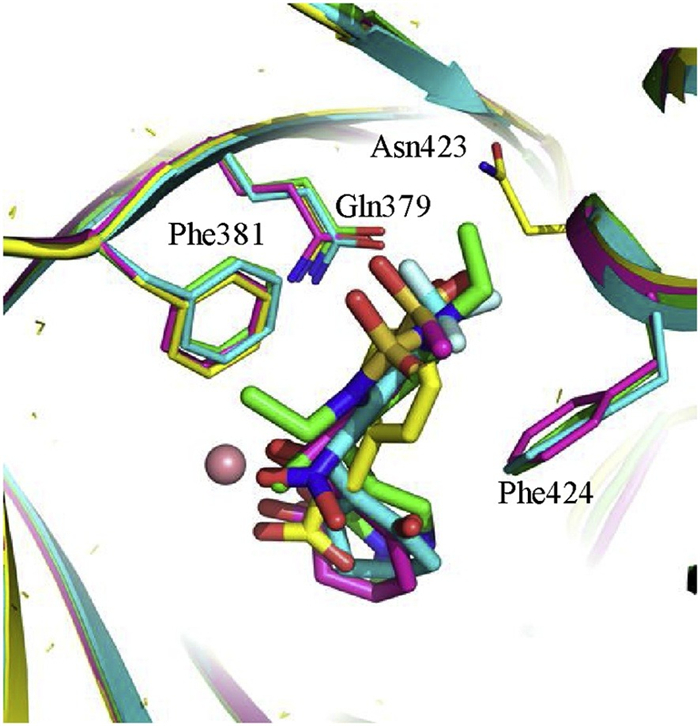
It is interesting to compare the HPPA binding mode in this study to the ones of known HPPD inhibitors [28] NTBC (PDB ID: 5CTO), sulcotrione (PDB ID: 5YWG) and Y13508 according to the crystal structures (Fig. 5). For each inhibitor in Fig. 5, two carbonyl groups form chelation interaction with the ferrous ion and its aromatic ring forms a π-π stacking interaction with side chains of Phe381 and Phe424. The overall binding mode of HPPA is quite like the binding modes of these three inhibitors, except that HPPA phenyl ring only forms a T-π stacking against the phenyl sidechain of Phe381. The minimum substructures of these HPPD inhibitors mimic the α-keto acid moiety of HPPA. The orientation of their aromatic rings resembles the HPPA phenyl ring. The QM/MM calculations combined with QM calculations predict that the free energy change for the binding of dioxygen to the iron is only -1.5 kcal/mol, suggesting a very weak negative binding affinity between them. As illustrated in previous section (Fig. 3), the phenyl ring will block the further association of dioxygen to the ketone carbon, hence the bound HPPA will behave like an inhibitor due to the failure of associating the O2. The aromatic rings of the HPPD inhibitors show similar shape and orientation to the HPPA phenyl ring. Following the story of the latter, these aromatic rings will also repel the association of dioxygen to the adjacent ketone carbons. Thus, these inhibitors exert their roles by preventing the addition of O2 and suppressing the oxidation of the active site metal ion. This inference is supported by the experimental observation that the HPPD does not bind O2 in the presence of its inhibitors.

|
Download:
|
| Fig. 5. Binding modes of the HPPA (yellow scheme), Y13508 (green scheme), NTBC (blue scheme) and sulcotrione (pink scheme) in the catalytic site of HPPD. | |
{kind=link}
In our experiments on the conversion reaction of HPPA catalyzed by AtHPPD, significant substrate-inhibition effect was also observed with Ksi =226.0 μmol/L and Km =44.4 μmol/L (see Supporting information for details). The inhibition constants (Ki) of NTBC [28], sulcotrione and Y13508 against HPPD were reported to be 6.43, 5.79 and 1.30 nmol/L, respectively. These data imply that HPPA binds to HPPD with different conformations. Some conformations lead to the reaction product HGA or the intermediate HPA. Some others do not react with HPPD leading to substrate self-inhibition. In this study, we prove that the substrate conformation observed in the AtHPPD-HPPA crystal structure is an inactive form, which contributes to the substrate self-inhibition of HPPD.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThe work was supported by the National Key R & D Program (No. 2018YFD0200100), National Natural Science Foundation of China (Nos. 21837001, 21273089, 22007035, U20A2038), the Open Project Fund of the Key Laboratory of the Pesticides and Chemical Biology of Central China Normal University (No. 2018-A01), the Fundamental Research Funds for the South-Central University for Nationalities (No. CZW20020), the Fundamental Research Funds for the Central Universities (No. KJ02072020-0657) and Hubei Province Natural Science Foundation (No. 2020CFB487).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.02.041.
| [1] |
N.P. Crouch, M.H. Lee, T. Iturriagagoitia-Bueno, et al., Cloning, Expression, and Purification of Mammalian 4-Hydroxyphenylpyruvate Dioxygenase/α-Ketoisocaproate Dioxygenase, Academic Press, Salt Lake City (2000). |
| [2] |
M. Kohlmeier, Amino Acids and Nitrogen Compounds, Academic Press, San Diego (2015). |
| [3] |
H. Ahrens, G. Lange, T. Muller, et al., Angew. Chem. Int. Ed. Eng. 52 (2013) 9388-9398. DOI:10.1002/anie.201302365 |
| [4] |
R. Beaudegnies, A.J. Edmunds, T.E. Fraser, et al., Bioorg. Med. Chem. 17 (2009) 4134-4152. DOI:10.1016/j.bmc.2009.03.015 |
| [5] |
D.W. Wang, H.Y. Lin, B. He, et al., J. Agric. Food Chem. 64 (2016) 8986-8993. DOI:10.1021/acs.jafc.6b04110 |
| [6] |
D.C. Bartlett, C. Lloyd, P.J. McKiernan, et al., J. Inherit. Metab. Dis. 37 (2014) 745-752. DOI:10.1007/s10545-014-9683-x |
| [7] |
D.D. Shah, J.A. Conrad, B. Heinz, et al., Biochemistry 50 (2011) 7694-7704. DOI:10.1021/bi2009344 |
| [8] |
D.D. Shah, J.A. Conrad, G.R. Moran, Biochemistry 52 (2013) 6097-6107. DOI:10.1021/bi400534q |
| [9] |
T. Borowski, A. Bassan, P.E. Siegbahn, Biochemistry 43 (2004) 12331-12342. DOI:10.1021/bi049503y |
| [10] |
C. Raspail, M. Graindorge, Y. Moreau, et al., J. Biol. Chem. 286 (2011) 26061-26070. DOI:10.1074/jbc.M111.227595 |
| [11] |
A. Wójcik, E. Broclawik, P.E.M. Siegbahn, et al., J. Am. Chem. Soc. 136 (2014) 14472-14485. DOI:10.1021/ja506378u |
| [12] |
H.Y. Lin, X. Chen, J.N. Chen, et al., Research 2019 (2019) 2602414. |
| [13] |
J.F. Lin, Y.L. Sheih, T.C. Chang, et al., PLoS One 8 (2013) e69733. DOI:10.1371/journal.pone.0069733 |
| [14] |
M. Gunsior, J. Ravel, G.L. Challis, et al., Biochemistry 43 (2004) 663-674. DOI:10.1021/bi035762w |
| [15] |
T. Borowski, M. Quesne, M. Szaleniec, QM and QM/MM methods compared: case studies on reaction mechanisms of metalloenzymes, in: T. KarabenchevaChristova (Ed.), Advances in Protein Chemistry and Structural Biology, Academic Press, Salt Lake City, 2015.
|
| [16] |
K. Johnson-Winters, V.M. Purpero, M. Kavana, et al., Biochemistry 42 (2003) 2072-2080. DOI:10.1021/bi026499m |
| [17] |
G.R. Moran, Arch. Biochem. Biophys. 544 (2014) 58-68. DOI:10.1016/j.abb.2013.10.022 |
| [18] |
C. Yang, J.W. Pflugrath, D.L. Camper, et al., Biochemistry 43 (2004) 10414-10423. DOI:10.1021/bi049323o |
| [19] |
I.M. Fritze, L. Linden, J. Freigang, et al., Plant Physiol. 134 (2004) 1388-1400. DOI:10.1104/pp.103.034082 |
| [20] |
J.M. Brownlee, K. Johnson-Winters, D.H.T. Harrison, et al., Biochemistry 43 (2004) 6370-6377. DOI:10.1021/bi049317s |
| [21] |
I.M. Fritze, L. Linden, J. Freigang, et al., Plant Physiol. 134 (2004) 1388-1400. DOI:10.1104/pp.103.034082 |
| [22] |
J. Brownlee, P. He, G.R. Moran, et al., Biochemistry 47 (2008) 2002-2013. DOI:10.1021/bi701438r |
| [23] |
A. Wójcik, M. Radon, T. Borowski, J. Phys. Chem. A 120 (2016) 1261-1274. DOI:10.1021/acs.jpca.5b12311 |
| [24] |
T. Borowski, A. Bassan, P.E.M. Siegbahn, Chem. Eur. J. 10 (2004) 1031-1041. DOI:10.1002/chem.200305306 |
| [25] |
S. Ye, C. Riplinger, A. Hansen, et al., Chemistry 18 (2012) 6555-6567. DOI:10.1002/chem.201102829 |
| [26] |
M.M. Mbughuni, M. Chakrabarti, J.A. Hayden, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 16788-16793. DOI:10.1073/pnas.1010015107 |
| [27] |
J.C. Price, E.W. Barr, B. Tirupati, et al., Biochemistry 42 (2003) 7497-7508. DOI:10.1021/bi030011f |
| [28] |
H.Y. Lin, J.F. Yang, D.W. Wang, et al., FEBS J. 286 (2019) 975-990. DOI:10.1111/febs.14747 |