 2021, Vol. 32
2021, Vol. 32 


Phosphorus containing compounds are of wide-ranging importance with applications in the development of organic synthesis [1], materials science [2] and medicinal chemistry [3]. As a privileged structure motif, 1, 2-oxaphosphole-3-ene 2-oxide are particularly attractive for the good biological properties such as antiviral activity, cytotoxicity and genotoxicity, and antimicrobial activity [4]. For the strategy on the construction of such a useful skeleton, it was reported that 1, 2-oxaphosphole-3-ene 2-oxide derivatives could be achieved through cyclization from the phosphorus-containing alkynes or allenes through Pd- or Rh-catalysis [5]. However, pre-introduction of phosphorus groups into the substrates made the method be far from the "ideal synthesis". Therefore, developing a sequential reaction from simple starting material to 1, 2-oxaphosphole-3-ene 2-oxide directly remains highly desirable.
Indole, as one class of alkaloids with high reactivity, exists in a wide range of natural products and bioactive molecules [6]. The design and construction indole-based compounds which may bring new or increased bioactivities are of great significance. Recently, 3-indolymethanols has been widely utilized as platform molecules to build indole-based heterocycles [7]. In the presence of Brønsted acid or Lewis acid, 3-indolymethanols which can act as Michael addition acceptors easily dehydrate to generate carbocation intermediates (Ⅰ) in situ, which is a resonance structure of vinyliminium (Ⅱ). The intermediate can be trapped easily by nucleophiles via 1, 2-additon or 1, 4-addition leading to C2-functionalized indoles or C3-functionalized indoles (Scheme 1-1) [8-10]. Comparing to 1, 4-addition, the 1, 2-addition of the intermediate have been rarely reported. Shi and Chen's groups reported three examples to construct C2-functionalized indoles [10]. Based on this strategy, chiral indole derivatives were also synthesized in acceptable yields with high enantioselectivity under acidic conditions [11]. When a reaction partner bearing both an electrophilic site and a nucleophilic site is employed, the intermediate (Ⅱ) can experience a 1, 4-addition and [3 + n] cycloaddition, affording indole-fused cyclic frameworks (Scheme 1-2) [12]. Although the significant progress has been achieved in the reaction chemistry of 3-indolymethanols, new reaction mode was seldom reported. We are eager to know if hydroxyl of 3-indolymethanol could be retained which act as four-atom building blocks to participate in the [4 +n] cyclization reaction in the presence of acid catalyst, which would lead to indole fused O-heterocycles those have attracted the attention of synthetic chemists because of their good biological activities in anti-tumor and treatment of arthritis [13].

|
Download:
|
| Scheme 1. Transformation of 3-indolymethanols promoted by acids. | |

|
Download:
|
| Scheme 2. Substrate scope of phosphite and 3-indolylmethanol. Reaction conditions: 1 (0.2 mmol), 2 (1.0 mmol), Mn(OAc)3·2H2O (0.8 mmol), DCM (2 mL), at 80℃ under N2. The dr values are determined by 31P NMR analysis. | |
In recent years, we have been interested in the transformation of indolylmethanols, a series of new reaction modes were discovered [14]. As an ongoing work, herein, we report a site-selective phosphonyl radical addition-cyclization relay reaction (Scheme 1-3). In this reaction, manganese triacetate serves as an oxidant for the generation of phosphonyl radical and a source for acetic acid release which resulted in the intramolecular transesterification reaction.

|
Download:
|
| Scheme 3. Gram-scale synthesis and further transformation. | |
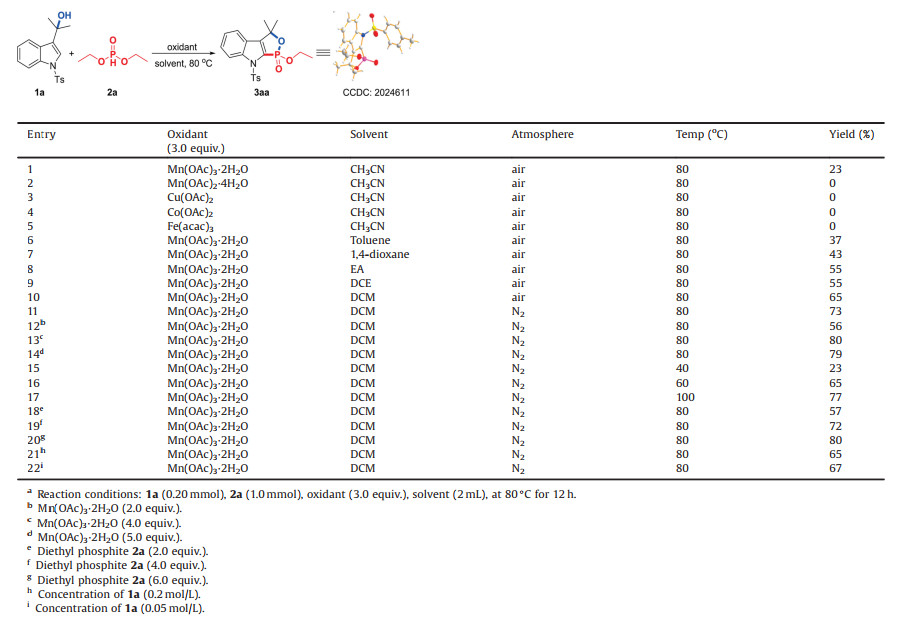
We evaluate our approach by examining the model reaction of 2-(1-tosyl-1H-indol-3-yl)propan-2-ol (1a) with diethyl phosphite (2a) (Table 1). Fortunately, by using Mn(OAc)3·2H2O as oxidant in acetonitrile under air atmosphere at 80℃, the reaction afforded the 1, 2-oxaphospholoindole derivative (3aa) in 23% yield (Table 1, entry 1). The definite structure of 3aa was determined by X-ray diffraction analysis. The type of oxidant was critical for the reaction efficiency, and it was found that Mn(OAc)3·2H2O could promote the formation of 3aa (Table 1, entries 1-5). Of the solvents tested, dichloromethane (DCM) was most effective for this transformation (Table 1, entries 6-10). When the reaction was carried out at the nitrogen atmosphere, the yield of the desired product was increased to 73% (Table 1, entry 11). Among the amount of manganese triacetate screened, 4.0 equiv. Mn(OAc)3·2H2O gave the highest yield (Table 1, entries 12-14). However, variation of reaction temperature (Table 1, entries 15-17), the equivalent of reagent 2a (Table 1, entries 18-20) and the concentration of 1a (Table 1, entries 21, 22) could not improve the reaction further. After a systematic screening, the desired product 3aa was obtained in 80% yield under the optimal conditions (Table 1, entry 13).
|
|
Table 1 Optimization of the reaction conditions.a |
{kind=link}
{kind=link}
{kind=link}
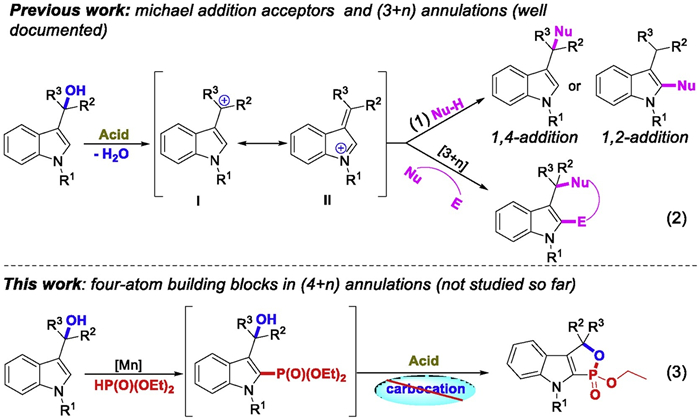
With the optimal conditions in hand, we evaluated the scope of this reaction (Scheme 2). Initially, we were pleased to observe that diethyl, dimethyl, di-n-butyl, diisopropyl and dibenzyl phosphites could lead to corresponding products in 68%-80% yields (3aa-3ae). To our delight, the ethyl phenylphosphinate and enthyl methylphosphinate, which have rarely been applied in radical reaction, furnished the desired products (3af-3ag, 63%-67%). In contrast, when diphenyl phosphite was investigated, the desired product 3ah could not be isolated. Furthermore, the substituents on the benzene ring in the indole motif including electron-donating groups (Me, OMe), halogens (F, Cl, Br) and electron-withdrawing groups (Ac, CF3) were also studied (3ba-3la, 45%-84%). Besides the electronic effect of various functional groups, the position of the substituents on the benzene ring also has great influence on the reaction outcomes (3ca, 3fa, 3ga). When two ethyl groups in side-chain were explored instead of two methyl groups, the transformation could lead to the desired product 3ma in moderate yield. In contrast, the diaryl-substituted 3-indolylmethanols 1n proved to be unsuccessful for the reaction which might be due to the too active substrate. Substrates bearing two different groups at the α-position of hydroxyl were also participated in the relay reaction smoothly and gave the products 3oa (63%), 3pa (36%) and 3pa' (30%) in moderate yields. In addition, the trans relationship between the methyl and the ethoxy was solved through X-ray crystallographic analysis of compound 3pa'. Intriguingly, secondary alcohol which was applied in lieu of a tertiary one led to 1, 2-oxaphospholoindoles 3qa and 3ra in 60% and 61% yield, respectively, while a lower yield was obtained with a primary alcohol permitted (3sa, 20%). The low yield for 3w is rationalized by DFT calculations. The predicted ΔG≠ for the intramolecular nucleophilic cyclization of INT3b derived from 1w (34.8 kcal/mol) is 7.6 kcal/mol higher than that of INT3 derived from 1a, suggesting that the formation of 3w is much less favorable than that of 3a (Fig. S2 in Supporting information). Geometrical inspection for INT3 indicates that the presence of two methyl groups could lead to steric hindrance with C4-H moiety of indole skeleton. Consequently, the rotation of the C3-C1 bond is constrained and the O···P distance in INT3 is 2.98 Å. However, for INT3b, such steric hindrance is negligible due to the absence of two methyl groups and therefore the distance of O···P increases to 3.51 Å. Thus, the formation of 3w is not ready to occur and low yield is obtained experimentally. Finally, pyrimidinyl group as protecting group on the indole N-1 position was also tolerated under the mild reaction condition (3ta, 73%). Unfortunately, N-CH3-3-indolylmethanol and isatin-derived 3-indolylmethanol were not competent substrates (3ua, 3va).
In order to test the gram-scale applicability of this transformation, 1a (4 mmol) was introduced into the reaction under the standard conditions. The reaction afforded the 1, 2-oxaphospholoindole derivative 3aa in a slightly reduced yield (72%). Further derivation of 3aa could lead to the synthesis of 1, 2-oxaphosphole 2-oxide 4a in 84% yield (Scheme 3).
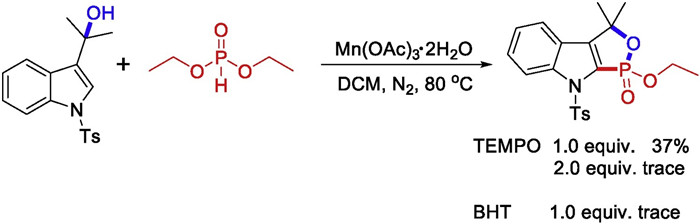
To shed light on the possible mechanism of the radical transformation, some control experiments were carried out (Scheme 4). With 1 equiv. of radical scavenger 2, 2, 6, 6-tetramethylpiperdin-1-oxyl (TEMPO) added, the product 3aa was obtained in a lower yield. When the 2 equiv. TEMPO or 1 equiv. 2, 6-di-tert-butyl-4-methylphenol (BHT) was introduced, the reaction was completely suppressed, suggesting that radical process might be involved in this reaction.

|
Download:
|
| Scheme 4. Control experiment. | |
{kind=link}
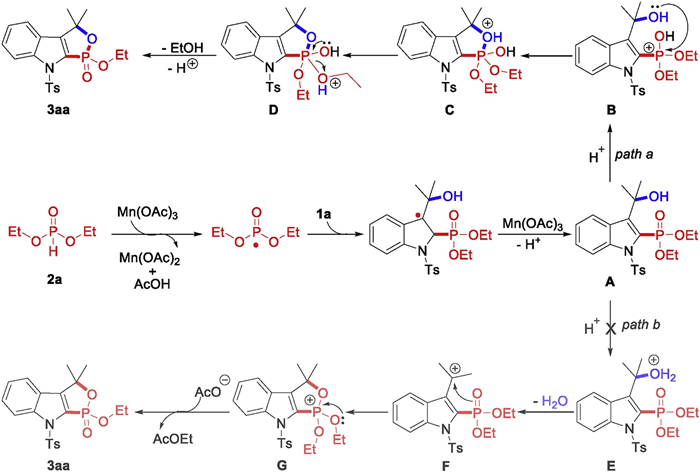
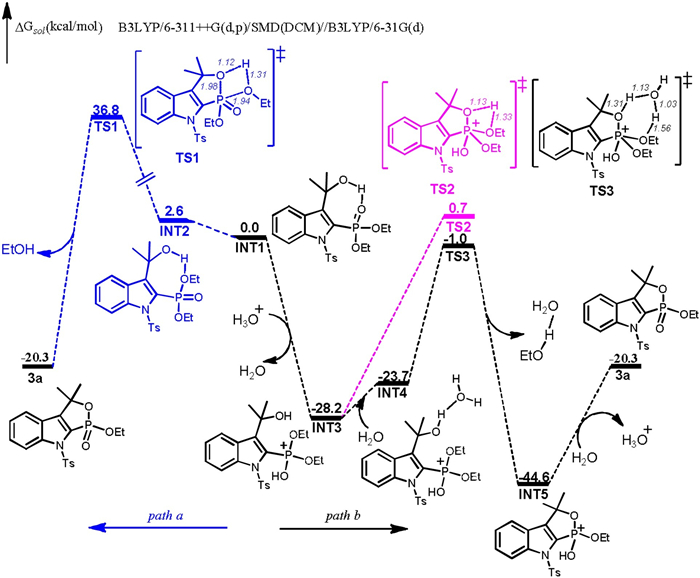
Inspired by the control experiments and previous reports [15], a plausible mechanism of this transformation was depicted inScheme 5. Initially, 2a could be converted to a phosphonyl radical by the oxidation of Mn(OAc)3·2H2O. Subsequently, the formed phosphonyl radical could attack C2 of 3-indolymethanols to yield a radical adduct, which could further undergo deprotonation to afford intermediate A. An intramolecular H-bonding interaction O-H···O is found in the intermediate A according to DFT calculations. Next, a possible direct intramolecular nucleophilic cyclization of A to afford the desired product 3aa was considered computationally (path a, Fig. 1). The corresponding transition state (TS) is located as TS1, in which the O···P distance is shortened to 1.98 Å along with the trend of dissociation of EtOH. The predicted ΔG≠ for direct cyclization is 34.2 kcal/mol, suggesting that this step is not ready to occur. One may suggest that in situ generated HOAc might assist the intramolecular nucleophilic cyclization of A. This possibility was also considered. However, even higher activation barrier is found for this cyclization step when a HOAc molecule is present H-bonding with A. Alternatively, one may propose that the hydroxyl group of A could be protonated and hence to afford a cation after dehydration. Interestingly, computational results suggest that O atom of P=O moiety is more ready to be protonated than the hydroxyl group and thus INT3 is formed (path b, Fig. 1). Afterward, the aforementioned intramolecular nucleophilic cyclization is considered and the corresponding TS is shown as TS2. The predicted ΔG≠ for the cyclization of INT3 is 28.9 kcal/mol, which is substantially lowered than the direct cyclization of A. The protonated phosphite group becomes more electrophilic and thus the cyclization of INT3 is ready to occur. When a water molecule is present and H-bonding with INT3, the calculated activation barrier decreases further (27.2 kcal/mol). Finally, the deprotonation of the formed INT5 is followed to yield the product 3aa. According to the computational analysis, a plausible mechanistic pathway is depicted in Scheme 5. The phosphite cation (B), instead of carbon cation (F), is suggested to be the key intermediate in the subsequent cyclization to afford the desired product.

|
Download:
|
| Scheme 5. A plausible mechanism. | |
{kind=link}

|
Download:
|
| Fig. 1. Energy profiles for the formation of product 3a. Bond lengths are shown in Å. | |
{kind=link}
In conclusion, we have demonstrated a novel relay strategy that Mn(Ⅲ) promoted radical addition-intramolecular transesterification of 3-indolylmethanols with phosphites for the synthesis of 1, 2-oxaphospholoindole derivatives under mild conditions. In the transformation, 3-indolylmethanols have been utilized as four-atom building blocks in [4+1] annulations which have scarcely been reported. Further studies on expanding the new reaction mode of 3-indolylmethanols are currently in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the National Natural Science Foundation of China (Nos. 21772138 and 21672157), the project of scientific and technologic infrastructure of Suzhou (No. SZS201708) and PAPD.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.02.001.
| [1] |
(a) W.J. Tang, X.M. Zhang, Chem. Rev. 103 (2003) 3029-3070; (b) H.A. McManus, P.J. Guiry, Chem. Rev. 104 (2004) 4151-4202; (c) S.F. Zhu, Q.L. Zhou, Acc. Chem. Res. 50 (2017) 988-1001; (d) J.M. John, S.H. Bergens, Angew. Chem. Int. Ed. 50 (2011) 10377-10380; (e) W.L. Yang, H. Wang, Z.R. Pan, Z. Li, W.P. Deng, Chin. Chem. Lett. 31 (2020) 721-724. |
| [2] |
(a) C. Queffélec, M. Petit, P. Janvier, D.A. Knight, B. Bujoli, Chem. Rev. 112 (2012) 3777-3807; (b) J.L. Montchamp, Acc. Chem. Res. 47 (2014) 77-87; (c) S.O. Jeon, J.Y. Lee, J. Mater. Chem. 22 (2012) 7239-7244. |
| [3] |
(a) E.D. Clercq, Med. Res. Rev. 31 (2011) 118-160; (b) F.R. Alexandre, A. Amador, S. Bot, et al., J. Med. Chem. 54 (2011) 392-395; (c) X.J. Zhou, R.C. Garner, S. Nicholson, C.J. Kissling, D. Mayers, J. Clin. Pharmacol. 49 (2009) 1408-1416. |
| [4] |
(a) P.G. Syam, B.B. Hari, R.C. Naga, C.D. Reddy, S. Reddy, Synth. Commun. 39 (2009) 1310-1316; (b) K. Vanya, D. Asya, K. Karamfil, E. Dobromir, Genetika 41 (2009) 179-188; (c) V.V. Belakhov, M.A. Shneider, E.V. Komarov, B.I. Ionin, Khim. Farm. Zh. 22 (1988) 719-724. |
| [5] |
(a) F. Yu, X.D. Lian, S.M. Ma, Org. Lett. 9 (2007) 1703-1706; (b) Y. Lv, G.B. Hu, D.M. Ma, et al., J. Org. Chem. 80 (2015) 6908-6914; (c) I. Essid, C. Laborde, F. Legros, et al., Org. Lett. 19 (2017) 1882-1885; (d) T. Ryu, J. Kim, Y. Park, S. Kim, P.H. Lee, Org. Lett. 15 (2013) 3986-3989; (e) D. Eom, Y. Jeong, Y.R. Kim, et al., Org. Lett. 15 (2013) 5210-5213. |
| [6] |
(a) C.X. Zhuo, C. Zheng, S.L. You, Acc. Chem. Res. 47 (2014) 2558-2573; (b) R.J. Phipps, N.P. Grimster, M.J. Gaunt, J. Am. Chem. Soc. 130 (2008) 8172-8174; (c) S.D. Yang, C.L. Sun, Z. Fang, et al., Angew. Chem. Int. Ed. 47 (2008) 1473-1476; (d) Z.Z. Shi, S.T. Ding, Y.X. Cui, N. Jiao, Angew. Chem. Int. Ed. 48 (2009) 7895-7898; (e) X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Angew. Chem. Int. Ed. 48 (2009) 5094-5115; (f) E.F. Huang, L. Zhang, C.Y. Xiao, et al., Chin. Chem. Lett. 30 (2019) 2157-2159; (g) T.D. Tan, X.Q. Zhu, M. Jia, et al., Chin. Chem. Lett. 31 (2020) 1309-1312. |
| [7] |
(a) Y.C. Zhang, F. Jiang, F. Shi, Acc. Chem. Res. 53 (2020) 425-446; (b) G.J. Mei, F. Shi, J. Org. Chem. 82 (2017) 7695-7707; (c) L. Wang, Y.Y. Chen, J. Xiao, Asian J. Org. Chem. 3 (2014) 1036-1052; (d) X.P. Han, H. Li, R.P. Hughes, J. Wu, Angew. Chem. Int. Ed. 51 (2012) 10390-10393; (e) Y.C. Xiao, Q.Q. Zhou, L. Dong, T.Y. Liu, Y.C. Chen, Org. Lett. 14 (2012) 5940-5943; (f) H. Wen, L. Wang, L.B. Xu, et al., Adv. Synth. Catal. 357 (2015) 4023-4030; (g) J. Xiao, H. Wen, L. Wang, et al., Green. Chem. 18 (2016) 1032-1037; (h) Y.Z. Zou, C.L. Chen, X.X. Chen, X.X. Zhang, W.D. Rao, Eur. J. Org. Chem. 2017 (2017) 2266-2271. |
| [8] |
N.A. Kogan, G.N. Kulbitskii, Chem. Heterocycl. Compd. 14 (1978) 46-48. DOI:10.1007/BF00635941 |
| [9] |
(a) K.T. Potts, J. Org. Chem. 26 (1961) 4719-4721; (b) X.F. Zeng, S.J. Ji, S.Y. Wang, Tetrahedron 61 (2005) 10235-10241; (c) J. Huang, G.X. Li, G.B. Yang, J.Z. Zhao, Z. Tang, Asian J. Org. Chem. 6 (2017) 1741-1744; (d) M.Y. Xiao, D.D. Ren, L.B. Xu, et al., Org. Lett. 19 (2017) 5724-5727; (e) J.A. Mackay, R.L. Bishop, V.H. Rawal, Org. Lett. 7 (2005) 3421-3424; (f) L.J. Zhou, Y.C. Zhang, J.J. Zhao, F. Shi, S.J. Tu, J. Org. Chem. 79 (2014) 10390-10398. |
| [10] |
(a) Y.X. Zou, X.Y. Liu, J. Zhang, et al., Adv. Synth. Catal. 361 (2019) 5311-5316; (b) X. Li, W. Tan, Y.X. Gong, F. Shi, J. Org. Chem. 80 (2015) 1841-1848; (c) Y. Liu, R.Y. Zhu, H.Z. Li, Y. Jiang, F. Shi, Synthesis 47 (2015) 1436-1446. |
| [11] |
(a) M. Rueping, B.J. Nachtsheim, S.A. Moreth, Angew. Chem. Int. Ed. 47 (2008) 593-596; (b) C. Guo, J. Song, J.Z. Huang, et al., Angew. Chem. Int. Ed. 51 (2012) 1046-1050; (c) C. Ma, F. Jiang, F.T. Sheng, et al., Angew. Chem. Int. Ed. 58 (2019) 3014-3020; (d) L. Song, Q.X. Guo, X.C. Li, J. Tian, Y.G. Peng, Angew. Chem. Int. Ed. 51 (2012) 1899-1902; (e) Y. Liu, H.H. Zhang, Y.C. Zhang, et al., Chem. Commun. 50 (2014) 12054-12057; (f) F.L. Sun, M. Zeng, Q. Gu, S.L. You, Chem. Eur. J. 15 (2019) 8709-8712. |
| [12] |
(a) B. Xu, Z.L. Guo, W.Y. Jin, et al., Angew. Chem. Int. Ed. 51 (2012) 1059-1062; (b) W. Tan, X. Li, Y.X. Gong, M.D. Ge, F. Shi, Chem. Commun. 50 (2014) 15901-15904; (c) F. Shi, H.H. Zhang, X.X. Sun, et al., Chem. Eur. J. 21 (2015) 3465-3471. |
| [13] |
(a) P. Malik, R. Bhushan, New J. Chem. 41 (2017) 13681-13691; (b) H.M. Li, K.M. Belyk, J.J. Yin, et al., J. Am. Chem. Soc. 137 (2015) 13728-13731; (c) J.L. Wood, B.M. Stoltz, S.N. Goodman, K. Onwueme, J. Am. Chem. Soc. 119 (1997)96529661. |
| [14] |
(a) Y. Zhou, W.B. Cao, L.L. Zhang, X.P. Xu, S.J. Ji, J. Org. Chem. 83 (2018) 6056-6065; (b) Y. Zhou, X.P. Xu, S.J. Ji, Org. Lett. 21 (2019) 2039-2042; (c) R.Y. Zhang, M.M. Xu, H.Y. Li, X.P. Xu, S.J. Ji, Chin. Chem. Lett. 32 (2021) 433-436. |
| [15] |
(a) T. Kagayama, A. Nakano, S. Sakaguchi, Y. Ishil, Org. Lett. 8 (2006) 407-409; (b) D. Ryzhakov, M. Jarret, J.P. Baltaze, et al., Org. Lett. 21 (2019) 4986-4990; (c) Y.Z. Gao, X.Q. Li, J. Xu, et al., Chem. Commun. 51 (2015) 1605-1607; (d) Y.Z. Gao, J. Wu, J. Xu, et al., RSC Adv. 4 (2014) 51776-51779; (e) J.F. Xue, S.F. Zhou, Y.Y. Liu, et al., Org. Biomol. Chem. 13 (2015) 4896-4902; (f) L. Li, J.J. Wang, G.W. Wang, J. Org. Chem. 81 (2016) 5433-5439; (g) M. Mondal, U. Bora, RSC Adv. 3 (2013) 18716-18754; (h) I.S. Hong, J. Suh, Org. Lett. 2 (2000) 377-380; (i) W.C. Yang, B. Li, M.M. Zhang, et al., Chin. Chem. Lett. 31 (2020) 1313-1316. |