 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory of Southwestern Chinese Medicine Resources, Hospital of Chengdu University of Traditional Chinese Medicine, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China
Organometallic chemistry based on homogeneous catalysis is an active research area in medicinal chemistry, since organic metal complexes act as chemically and biologically active molecules with a peculiar chemical structure and diverse evidence of efficacy [1]. Thanks to the successful use of Pt coordination complexes in oncotherapy, several organometallic frameworks have been designed and synthesized for the development of novel bioactive molecules in drug discovery. Among these scaffolds, those based on ferrocene have been in high demands due to the inherent stability of the sandwich-like ferrocene moiety to air, heat and light, and for presenting low toxicity, low cost and reversible redox properties without environmental pollutions and physical hazards [2-5].
Numerous strategies have been reported to construct ferrocene containing bioactive pharmacophores that can act as anti-proliferative, anti-plasmodial, anti-microbial, anti-inflammatory, antioxidant, anti-tubercular, among others. Among these broad therapeutic potentials, their antineoplastic properties are notorious, as the metabolic formation of ferrocenium can trigger accumulation of reactive oxygen species (ROS), which leads to DNA oxidative damage. The ferrocene moiety has been introduced into several drug-like scaffolds, such as pyrazol, N-heterocyclic carbine, 1H-1, 2, 3-triazole, selenourea, nucleobase and indeno[1, 2-C] isoquinolines, as well as into natural products, such as podophyllotoxin, biotin, cinchona, uracil and estrogen, to obtain antitumor hybrid molecules (Fig. 1A) [6-11]. However, ferrocene insertion strategies rarely focus on the drug skeleton for the design of new derivatives of spirooxindole, a classical anticancer drug [12]. Furthermore, although the achiral and racemic ferrocene derivatives account for the most of the new biologically screened compounds, little stereoselective strategy has been applied in this area.

|
Download:
|
| Fig. 1. (A) Selected anti-cancer ferrocene containing molecules derived from privileged scaffolds and natural products; (B) C3-spirooxindole derived natural product and synthetic molecular reported to inhibit MDM2–p53 interaction; (C) Rational drug design based on preliminary docking study and structure–activity relationships. | |
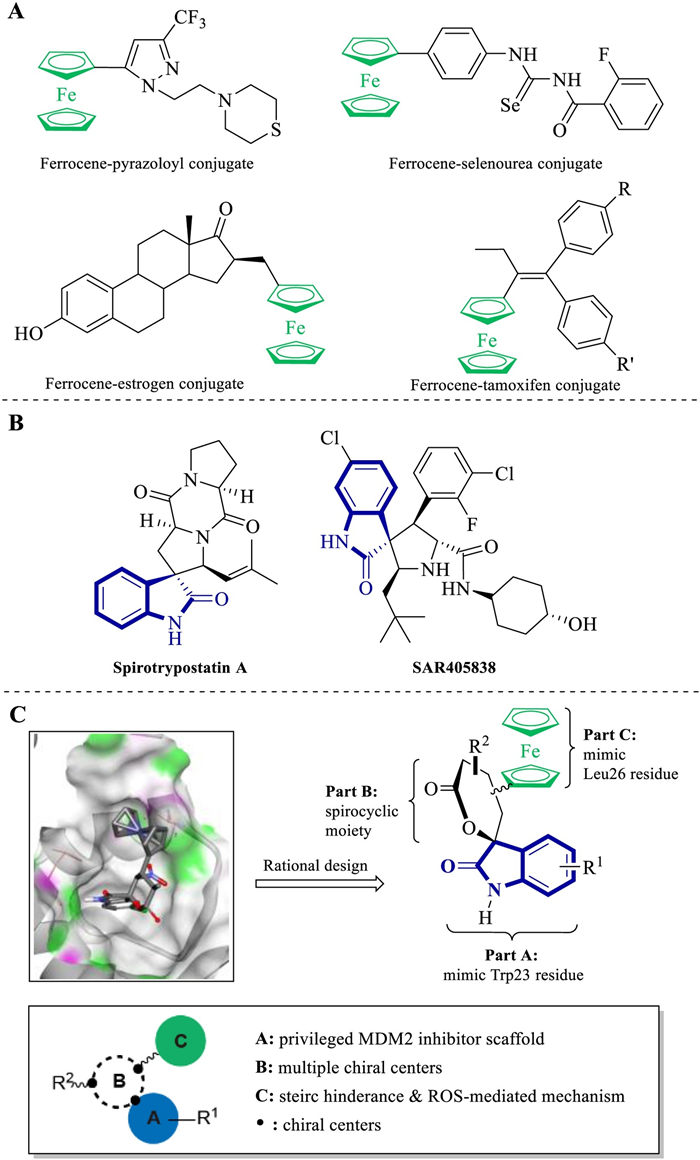{kind=link}
P53, also called "the guardian of the genome", is a tumor suppressor protein that can effectively regulate the cell cycle, apoptosis, DNA repair and senescence. About half of human cancers, such as sarcomas, gliomas, hematological malignancies, melanomas and carcinomas, express mutant p53, which becomes inactive. Meanwhile, in the other half of the tumors that conserve wild-type p53, it becomes non-functional when interacting with the MDM2 protein [13]. The down-regulation of MDM2–p53 protein–protein interaction was considered a novel therapeutic approach for the tumor patients [14]. Thus, the design and synthesis of small-molecule MDM2 inhibitors derived from privileged scaffolds, especially the C3-spirooxindole skeleton, attracted several chemists and pharmacologists for targeting the discovery of anticarcinogens (Fig. 1B) [15-20].
The analysis of the p53–MDM2 complex crystal structure revealed that the binding of p53 to MDM2 was mediated mainly by three hydrophobic residues (Phe19, Trp23 and Leu26) of p53 and a deep hydrophobic pocket in MDM2. As the C3-spirooxindole was identified as a perfect Trp23 mimic, computational docking studies suggested that the introduction of the ferrocene group into the spirocycle could facilitate a better adjustment of this scaffold within the MDM2 hydrophobic cleft. The ferrocene moiety could play a dual role in increasing affinity for MDM2 and triggering ROS accumulation with its redox properties.
Continuing our interest in the construction of relevant medical scaffolds using stereoselective organocatalysis and ferrocene-based rational molecular drug design (Fig. 1C) [21-24], we envisaged an organocatalytic multicomponent organocatalytic tandem reaction [25, 26]. Thus, we were able to asymmetrically produce a library of chiral oxa–spirooxindole–ferrocene hybrids bearing various stereogenic centers and functional groups [27-29]. All our products obtained fortunately comply with the Lipinski's rules of five parts from molecular overweight caused by ferrocenyl, and their clogP values ranged from 3.2–4.7. This chiral ferrocene heterocycle library can lay a solid foundation for developing novel and efficient MDM2 inhibitors.
Employment of ferrocenyl-substitued nitroalkenes, saturated aldehydes and isatins generated the desired products 5a–5m with four contiguous stereocenters in good yields with excellent diastereo- (up to > 20:1 dr) and enantio- (up to > 99% ee) selectivities (Scheme 1, see Table S1 in Supporting information for screening of reaction conditions). The nature and position of substitution on isatins slightly affected the reaction (5a–5k). Using n-butyraldehyde and isovaleraldehyde generated additional diversity at the R2 position, allowing the production of 5l and 5m. The chiral spiro hemiacetals 4 could also be converted into spirooxindole tetrahydropyrans 6 and spirooxindole 3, 4-dihydropyrans 7; yields were good, diastereoselectivity and enantioselectivity were outstanding (> 20:1 dr, up to > 99% ee). The absolute configuration of the resulting spirooxindoles 5, 6 and 7 was assigned based on X-ray crystallographic analysis of 4d (Scheme 1).

|
Download:
|
| Scheme 1. Asymmetirc synthesis of chiral spirocyclic oxindole-ferrocene hybrids. Reactions were performed with 2 (0.4 mmol), 3 (0.36 mmol), Hayashi-Jørgensen secondary amine catalyst (0.04 mmol) and AcOH (0.04 mmol) in 2mL CH2Cl2 at r.t. for 3 h, after which 1 (0.2 mmol) and TABA (0.02 mmol) were added. Oxidation of 4 with DMP generated spirooxindole δ-lactone 5. Reduction of 4 with Et3SiH and BF3·Et2O at −20 ℃ generated spirooxindole tetrahydropyran 6. Protonation of 4 with para-toluenesulfonic acid at 40 ℃ generated spirooxindole 3, 4-dihydropyran 7. Yields were calculated from the isolated pure diastereomer; dr values, from 1H NMR analysis of the crude reaction mixture; and ee values, from chiral HPLC analysis. | |
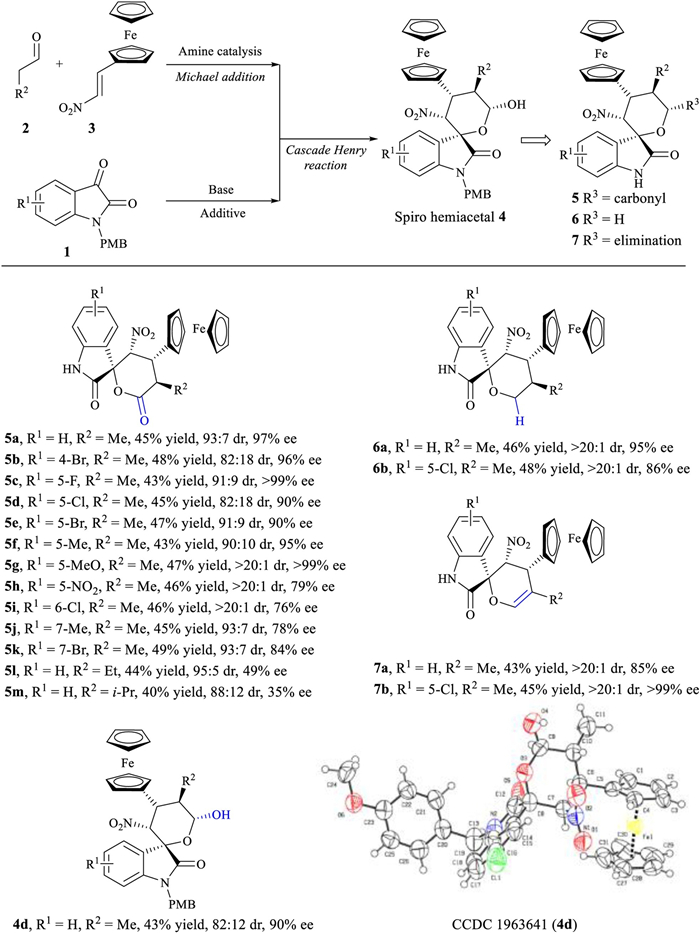{kind=link}
From this series of chiral, bioorganometallic, ferrocenyl-containing spirooxindoles, we determined their inhibitory capacities on MDM2 and their cytotoxicity on breast cancer cells with different p53-mutation status (Fig. 2 and Tables S1–S3 in Supporting information). As shown in Fig. 2A, almost all compounds showed moderate to negligible cellular proliferation inhibitory effects on p53-mutated MDA-MB231 cells. The exceptions where 5c–5g and 5i compounds, which showed better abilities to bind to the MDM2 protein, in addition to significantly inhibiting the proliferation of p53-wild MCF-7 and ZR-75−1 cells. The bioactivities of synthetic compounds were mainly influenced by the substitution of R1 group in the isatin scaffold. Halogen substitution at the 5- or 6-position of the isatin scaffold maintained the inhibitory capacities on both MDM2 and p53-wild cells proliferation. The halogen group substitution by a nitro group led to a significantly weaker inhibition of MDM2 and cell proliferation. The alkyl substitution in the R2 position demonstrated minor influences on the MDM2 binding, and among these substitutions, methyl group showed better inhibition effects than ethyl or iso-propyl groups. The transformation of carbonyl group into methylene or unsaturation vinyl groups resulted in the decline of MDM2 binding and breast cancer cells cytotoxicity. It is worth noting that p53-wild breast cancer cell lines (MCF-7 and ZR-75−1, blue in Fig. 2C) were significantly more sensitive to compound 5d than those containing p53-mutated forms (orange in Fig. 2C). The most sensitive cell line to 5d, with the IC50 value at submicromolar level, was selected to perform the subsequent studies.

|
Download:
|
| Fig. 2. In vitro bioassays. (A) The inhibitory activities of synthetic compounds (1.0 μmol/L) on MDM2 and proliferation of breast cancer cell lines; (B) The Ki values of selected compounds on MDM2 protein; (C) The IC50 values of compound 5d on a panel of breast cancer cell lines (blue: p53-wild, orange: p53-mutated); (D) The binding modes of 5d in MDM2; (E) The 2D depiction of 5d and its interacted residues; (F) and (G) The oxidative damage of ZR-75-1 cells induced by 5d were determined by DCFDA (F) and TrxR assays (G). Data are expressed as mean±SD (n=3, student's t-test). *P < 0.05, **P < 0.01vs. control. | |
{kind=link}
The potential binding modes of 5d, the most active compound found, to the p53 binding pocket of MDM2 were shown in Figs. 2D and E. Molecular docking was performed using the CDOCKER method (embedded in the Accelrys Discovery Studio 3.5 package) and the MDM2 protein structure was downloaded from the protein data bank (PDB) database (https://www.rcsb.org, PDB ID 4LWU). Protein preparation and molecular docking were performed according to the manufacturer's protocols and default settings, without special statements. Figs. 2D and E displayed the binding mode of 5d to the active site of MDM2 for exerting antitumor activity [23, 30]. The binding conformations of 5d to MDM2 were shown in Fig. 2D. The accessible surface of binding pocket was contoured by its H-bond (hydrogen bond) donor or acceptor properties. Structural analysis of the MDM2-5d complex revealed that the isatin and ferrocene group mimiced the Trp23 and Leu26 residues of p53 to bind to the MDM2 protein (Fig. 2D). N1 atom of the isatin fragment formed stable hydrogen bonds to the Leu54 residue and the His96 residue established hydrophobic interactions with the ferrocene group (Fig. 2E). In addition, the ferrocene group helped 5d to further stabilize the inhibitor protein interactions on account of a large steric hindrance. The oxidative damage induced by 5d in ZR-75−1 cell was determined by the 2′, 7′-dichlorofluorescin diacetate (DCFDA) and thioredoxin reductase (TrxR) assays (Figs. 2F and G). After incubation with 5d compound, the fluorescence intensities of the ROS probe inside the DCFDA cells increased over time, with their maximum values being reached after 48 h of incubation. In addition, the intracellular TrxR activities decreased after 5d treatment in a dose-dependent manner. This suggests that 5d may validly interfere with the p53–MDM2 interaction as well.
Apoptotic cell death of ZR-75−1 cells induced by 5d compound was detected by an Annexin V-FITC/PI assay in a flow cytometry instrument (Fig. 3A) [31]. The average apoptosis positive rate of 1.0 and 5.0 μmol/L 5d were 9.2% and 19.5%, respectively. In addition, nuclei morphology changed in ZR-75−1 cells incubated with 5d were observed after Hoechst 33258 staining (Fig. 3B). We observed that there were more apoptotic nuclei fragments in the 5.0 μmol/L group than in the 1.0 μmol/L group, suggesting that 5d led to ZR-75−1 cell apoptosis in a dosage-dependent manner. Changes in expression of p53, MDM2, p21 proteins and apoptosis related markers (Bax, PARP and caspase-9) were characterized by protein immunoblotting (Fig. 3C). After incubation with 1.0 μmol/L of 5d compound for 24 h, the accumulations of p21 and p53 could be observed, although the MDM2 expression and PARP cleavage had not been noticeably changed. In the 5.0 μmol/L incubation group, the accumulations of p53, MDM2, p21 and Bax, as well as the cleavages of PARP and caspase-9 could be clearly observed in Western blot images. Above results suggested that 5d compound probably interfered MDM2-mediated p53 degradation, induced oxidative damage and apoptosis in ZR-75−1 breast cancer cells.

|
Download:
|
| Fig. 3. Compound 5d induced apoptosis in ZR-75-1 cells. (A) Apoptosis in ZR-75-1 cells incubated by 5d were quantitated by flow cytometry with Annexin V-FITC/propidium iodide dual staining; (B) The nuclei morphology changed in ZR-75-1cells after 5d incubation; (C) The expression levels changed of MDM2, p53, p21 and apoptosis related proteins after 5d incubation. | |
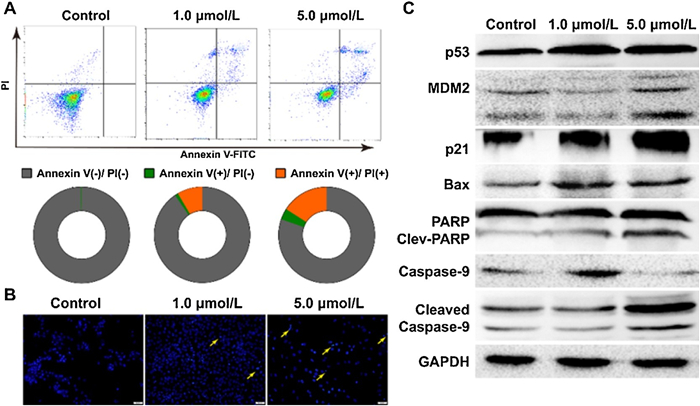{kind=link}
In conclusion, we have designed and synthesized a series of spirooxindole-ferrocene hybrids that combine a privileged scaffold for inhibiting MDM2 and a ROS-inducing lipophilic ferrocene group for the development of small molecule cancer therapeutics. A Michael–aldol–acetalization relay involving ferrocenyl-substitued nitroalkenes, saturated aldehydes and isatins were developed in order to obtain new molecules with therapeutical potentials, resulting in a collection of chiral, bioorganometallic, ferrocenyl-containing six-membered oxa-spirooxindoles with high yields and diastereoselectivities. Preliminary biological assays demonstrated that the most bioactive compound found (5d) suppressed p53-wild breast cancer cells proliferation and bound to MDM2 at submicromolar levels. In silico analysis suggested 5d compound interacted with MDM2 by mimicing the Trp23 and Leu26 residues of p53. The present work was able to provide some structural basis for the development of novel multifunctional MDM2 inhibitors. In addition, further exploration of more derivatives from this library and further organocatalysis application can be useful for the development of potential lead compounds in cancer targeted therapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was supported by grants from the National Natural Science Foundation (Nos. 21772131, 81672552, 81773890, 82073997), the Fundamental Research Funds of Science & Technology Department of Sichuan Province (Nos. 2019YFSY0004, 2017JY0226, 2019YFS0298) and Project First-Class Disciplines Development supported by CDUTCM.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.01.033.
| [1] |
G. Jaouen, A. Vessieres, S. Top, Chem. Soc. Rev. 44 (2015) 8802-8817. DOI:10.1039/C5CS00486A |
| [2] |
A.C. de Oliveira, E.A. Hillard, P. Pigeon, et al., Eur. J. Med. Chem. 46 (2011) 3778-3787. DOI:10.1016/j.ejmech.2011.05.043 |
| [3] |
M.A. Richard, D. Hamels, P. Pigeon, et al., ChemMedChem 10 (2015) 981-990. DOI:10.1002/cmdc.201500075 |
| [4] |
Y. Wang, P. Pigeon, S. Top, M.J. McGlinchey, G. Jaouen, Angew. Chem. Int. Ed. 54 (2015) 10230-10233. DOI:10.1002/anie.201503048 |
| [5] |
K. Wu, J.Y. Park, R. Al-Saadon, et al., Organometallics 37 (2018) 1910-1918. DOI:10.1021/acs.organomet.8b00186 |
| [6] |
X.H. He, Y.L. Ji, C. Peng, B. Han, Adv. Synth. Catal. 361 (2019) 1923-1957. DOI:10.1002/adsc.201801647 |
| [7] |
J.L. Li, Y.Q. Liu, W.L. Zou, et al., Angew. Chem. Int. Ed. 59 (2020) 1863-1870. DOI:10.1002/anie.201912450 |
| [8] |
X. Zhang, X. Li, J.L. Li, et al., Chem. Sci. 11 (2020) 2888-2894. DOI:10.1039/C9SC06377C |
| [9] |
W.J. Luo, B.X. Shao, J.Y. Li, et al., Org. Chem. Front. 7 (2020) 1016-1021. DOI:10.1039/D0QO00140F |
| [10] |
R.A. Hussain, A. Badshah, N. Ahmed, et al., Polyhedron 170 (2019) 12-24. DOI:10.1016/j.poly.2019.05.027 |
| [11] |
P. Chellan, P.J. Sadler, Chemistry 26 (2020) 8676-8688. DOI:10.1002/chem.201904699 |
| [12] |
W.H. Zhou, X.G. Xu, J. Li, et al., Chin. Chem. Lett. 28 (2017) 422-425. DOI:10.1016/j.cclet.2016.09.001 |
| [13] |
B. Yu, D.Q. Yu, H.M. Liu, Eur. J. Med. Chem. 97 (2015) 673-698. DOI:10.1016/j.ejmech.2014.06.056 |
| [14] |
Y.Q. Chen, J.J. Liang, T. Li, et al., Chin. Chem. Lett. 30 (2019) 924-928. DOI:10.1016/j.cclet.2019.02.013 |
| [15] |
S.P. He, G.Q. Dong, S.C. Wu, et al., J. Med. Chem. 61 (2018) 7245-7260. DOI:10.1021/acs.jmedchem.8b00664 |
| [16] |
C.J. Ji, S.Z. Wang, S.Q. Chen, et al., Bioorg. Med. Chem. 25 (2017) 5268-5277. DOI:10.1016/j.bmc.2017.07.049 |
| [17] |
T.T. Liu, Y. Jiang, Z.Z. Liu, et al., Medchemcomm. 8 (2017) 1668-1672. DOI:10.1039/C7MD00287D |
| [18] |
W. Liu, S.Q. Chen, F. Zhang, et al., Bioorg. Med. Chem. Lett. 29 (2019) 1636-1642. DOI:10.1016/j.bmcl.2019.04.037 |
| [19] |
S.Z. Wang, S.Q. Chen, Z.J. Guo, et al., Org. Biomol. Chem. 16 (2018) 625-634. DOI:10.1039/C7OB02726E |
| [20] |
S.Z. Wang, Y. Jiang, S.C. Wu, et al., Org. Lett. 18 (2016) 1028-1031. DOI:10.1021/acs.orglett.6b00155 |
| [21] |
H.P. Zhu, K. Xie, X.H. He, et al., Chem. Commun. (Camb.) 55 (2019) 11374-11377. DOI:10.1039/C9CC05916D |
| [22] |
F. Peng, Q. Zhao, W. Huang, et al., Green Chem. 21 (2019) 6179-6186. DOI:10.1039/C9GC02694K |
| [23] |
R. Zhou, Q.J. Wu, M.R. Guo, et al., Chem. Commun. (Camb.) 51 (2015) 13113-13116. DOI:10.1039/C5CC04968G |
| [24] |
B. Han, W. Huang, W. Ren, et al., Adv. Synth. Catal. 357 (2015) 561-568. DOI:10.1002/adsc.201400764 |
| [25] |
Y.H. Zhang, C.T. Wang, W. Huang, et al., Org. Chem. Front. 5 (2018) 2229-2233. DOI:10.1039/C8QO00422F |
| [26] |
Q. Zhao, C. Peng, H. Huang, et al., Chem. Commun. (Camb.) 54 (2018) 8359-8362. DOI:10.1039/C8CC04732D |
| [27] |
G.J. Mei, F. Shi, Chem. Commun. (Camb.) 54 (2018) 6607-6621. DOI:10.1039/C8CC02364F |
| [28] |
B. Yu, Z.Q. Yu, P.P. Qi, D.Q. Yu, H.M. Liu, Eur. J. Med. Chem. 95 (2015) 35-40. DOI:10.1016/j.ejmech.2015.03.020 |
| [29] |
L.M. Zhou, R.Y. Qu, G.F. Yang, Expert Opin, Drug Discov. 15 (2020) 603-625. |
| [30] |
Y. Zhao, A. Aguilar, D. Bernard, S. Wang, J. Med, Chem. 58 (2014) 1038-1052. |
| [31] |
B.W. Ke, M. Tian, J.J. Li, B. Liu, G. He, Med. Res. Rev. 36 (2016) 983-1035. DOI:10.1002/med.21398 |