 2021, Vol. 32
2021, Vol. 32 


2 Southern Laboratory of Ocean Science and Engineering (Guangdong, Zhuhai), Zhuhai 519000, China;
3 South China Sea Bio-Resource Exploitation and Utilization Collaborative Innovation Center, Guangzhou 510006, China;
4 State Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Open Laboratory of Applied Microbiology, Guangdong Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou 510070, China
Meroterpenoids, hybrid natural products of both nonterpenoid and terpenoid origin, can be classed into polyketide-, shikimate- and amino acid-derived (mainly indole-diterpenoids) meroterpenoids based on their biosynthetic pathways [1-4]. They are widely distributed in plants, animals, bacteria and fungi. Fungal meroterpenoids, as the talented representative, displayed rich structural diversity and widespread bioactivity [1-4], especially providing some clinically used drugs or promising leads exemplified by mycophenolic acid (its derivative used as an immunosuppressant drug) [5], fumagillin (the antimicrobial and anti-angiogenesis agent) [6, 7], antroquinonol and 4-acetyl antroquinonol B (phase II clinical drug of anticancer, antihypercholesterolemia and antihyperlipidemia) [8] and pyripyropene A (acyl-CoA: cholesterol acyltransferase inhibitor) [9].
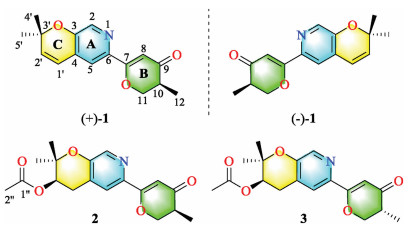
Marine-derived fungi are recognized as the essential sources of structurally unique and biologically active meroterpenoids [10, 11]. Our research group recently focuses on discovering novel secondary metabolites from ascidian-derived fungi [12-15], implying great potent chemical diversity and bioactivity. Herein, the fungusAmphichorda felina (A. felina) SYSU-MS7908, identified by the morphological and the internal transcribed spacer (ITS) of the nuclear ribosomal DNA data analysis, was isolated from a marine ascidian Styela plicata collected from North Atoll, South China Sea. Chemical investigation on the solid rice fermentation of this strain led to the isolation of four novel meroterpenoids, including a pair of enantiomers [amphichoterpenoid A, (±)-1], and two epimers (amphichoterpenoids B and C, 2 and 3) (Fig. 1). To our best knowledge, compounds 1–3 are the first case of picoline-derived meroterpenoids featuring a 6/6/6 tricyclic pyrano[3, 2-c]pyridinyl-γ-pyranone skeleton. Besides, the oil encapsulated nanodroplet crystallization (ENaCt) method [16] using a customized glass plate was applied to cultivating single crystals of 1 and 2 (Supporting information). Herein, the isolation, structure elucidation, biosynthetic pathway and biological activity of compounds 1–3 were described.

|
Download:
|
| Fig. 1. Chemical structures of (+/-)-1, 2 and 3. | |
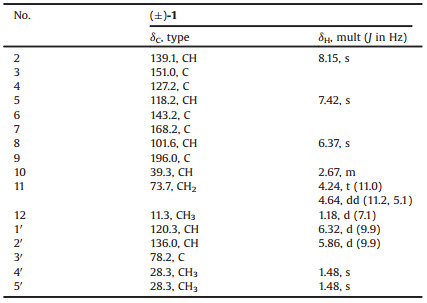
Amphichoterpenoid A (1) was obtained as a white solid, and its molecular formula was assigned to be C16H17NO3 by high resolution-electrospray ionization-mass spectrometry (HR-ESI-MS) data (m/z 272.1283 [M+H]+, calcd. 272.1287), indicating 9 degrees of unsaturation. The infra-red (IR) spectrum of 1 indicated the absorption of carbonyl (1658 cm-1) group. The 1H NMR spectrum (Table 1) revealed resonances corresponding to three olefinic protons [δH 6.32 (d, J = 9.9 Hz, H-1′); 5.86 (d, J = 9.9 Hz, H-2′); 6.37 (s, H-8)], two aromatic protons [δH 8.15 (s, H-2); 7.42 (s, H-5)] owing to a 3, 4, 6-trisubstituted pyridine ring, one methine [δH 2.67 (m, H-10)], one oxygenated methylene [δH 4.24 (t, J = 11.0 Hz, H-11a); 4.64 (dd, J=11.2, 5.1 Hz, H-11b)], and three methyls [δH 1.18 (d, J = 7.1 Hz, H-12); 1.48 (s, H-4′); 1.48 (s, H-5′)]. The 13C nuclear magnetic resonance (NMR) and distortionless enhancement by polarization transfer (DEPT) spectroscopy data (Table 1) of 1 showed the presence of 16 carbons, including six sp3 and ten sp2 hybridized carbons. Except for five sp2 hybridized carbons (δC 118.2, 127.2, 139.1, 143.2, 151.0) belonging to the pyridine ring, the remaining five sp2 hybridized carbons were sorted as one carbonyl group (δC 196.0) and two double bonds (δC 101.6, 168.2, 120.3, 136.0).
|
|
Table 1 1H (400 MHz) and 13C (100 MHz) NMR data of 1 in CDCl3. |
{kind=link}
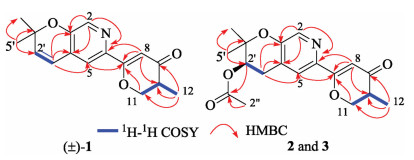
The planar structure of 1 was identified by the interpretation of 1H–1H correlation spectroscopy (1H–1H COSY) and heteronuclear multiple-bond correlation (HMBC) spectrum (Fig. 2). The core fragment of the pyridine ring (A) was established by the HMBC correlations from H-2 to C-3, C-4 and C-6, from H-5 to C-3, as well as the chemical shifts. The key HMBC correlations from H-5 to C-7; from H-8 to C-6, C-7 and C-10; from H-11 to C-7; and from H3-12 to C-9, C-10 and C-11, together with their chemical shifts, located a methyl dihydro-γ-pyranone ring moiety (B) at C-6 of the pyridine ring. The 1H–1H COSY correlation between H-1′ and H-2′ and the HMBC correlations from H-1′ to C-3, C-4 and C-5; from H3-4′ and H3-5′ to C-2′ and C-3′, along with the required degrees of unsaturation, revealed a dimethyl-substituted pyran ring (C) fused with the pyridine ring. Therefore, the tricyclic pyrano[3, 2-c]pyridinyl-γ-pyranone skeleton for 1 was established.

|
Download:
|
| Fig. 2. Key 1H-1H COSY and HMBC correlations of (±)-1, 2 and 3. | |
{kind=link}
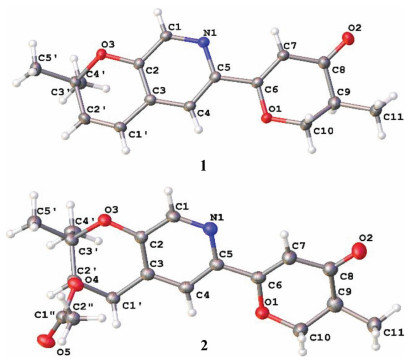
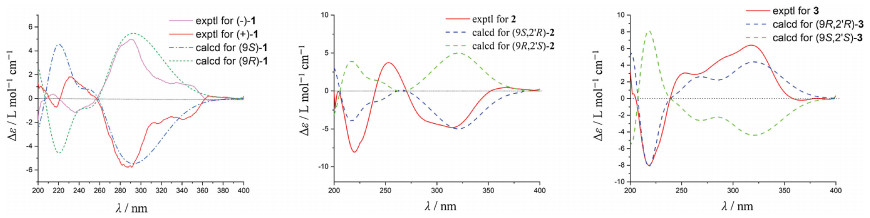
Compound 1 was crystallized from acetone using ENaCt method [16] to afford a single crystal. Follow-up X-ray crystallography confirmed the structure of 1 (Fig. 3). The P21/C space group of crystal structure suggested that compound 1 was a racemate, which agreed with the optical inactivity of its methanol solution. Subsequently, 1 was subjected to chiral HPLC separation to yield the enantiomers, (+)-1 (tR = 4.73 min, [α] +80.5) and (-)-1 (tR = 5.76 min, [α] -79.3). Their electronic circular dichroism (ECD) spectra displayed opposite Cotton effects at 292 nm (Fig. 4), deriving from n → π* transition of the dihydro-γ-pyranone chromophore. The absolute configuration of (+)/(-)-1 was deduced by comparing the experimental and calculated ECD spectra. The theoretical ECD spectra were calculated by a quantum chemical method at the (cam-b3lyp/def2svp) level, and the calculated ECD curve of (10R)-1 was comparable to the experimental one of (-)-1 (Fig. 4), suggesting the absolute configuration of (-)-1 as 10R. Thus, the structures of (+)-1 and (-)-1 were determined and named as (+)-(10S)-amphichoterpenoid A and (-)-(10R)-amphichoterpenoid A, respectively.

|
Download:
|
| Fig. 3. X-ray crystallographic analysis of (±)-1 and 2. | |
{kind=link}

|
Download:
|
| Fig. 4. Experimental and calculated ECD spectra of (+)/(-)-1, 2, and 3 (in MeOH). | |
{kind=link}
Amphichoterpenoids B (2) and C (3) had the same molecular formula of C18H21NO5 with nine unsaturation degrees based on the HR-ESIMS. Detailed analysis of their NMR spectroscopic data (Table S1 in Supporting information) suggested that 2 and 3 shared the same planar structure and were similar to 1 with the pyrano[3, 2-c]pyridinyl-γ-pyranone scaffold, except for the presence of additional signals attributed to an acetyl group (δC 170.5, C-1′'; δC 21.1, C-2′'; δH 2.06, s, H-2′'), one methylene and one methine (CH2-CH) (δC 28.0, C-1′; δH 2.82, dd, J=18.0, 4.4 Hz, H-1′a; δH 3.15, dd, J=18.0, 4.7 Hz, H-1′b; δC 69.8, C-2′; δH 5.05, t, J =4.6 Hz, H-2′) in 2 and 3. At the same time, the lack of two olefinic methines (CH=CH) [δH 6.32 (d, J = 9.9 Hz, H-1′); δC 120.3, C-1′; δH 5.86 (d, J = 9.9 Hz, H-2′); δC 136.0, C-2′] in 2 and 3 were observed. The planar structure of 2 and 3 (Fig. 1, Fig. 2) were established by the interpretation of two dimension (2D) NMR spectra (1H–1H COSY, HSQC and HMBC). The key HMBC correlations from H-2′ and H-2′' to C-1′' allowed assignment of the acetyl group at C-2′ in 2 and 3.
Compounds 2 and 3 were purified from the same fraction by the normal phase HPLC [97% n-hexane-isopropanol, tR (2) = 28.5 min, tR (3) = 25.5 min] with different optical rotations. The observed minor variation (± 0.01) of the chemical shifts of H-10, H-11 and H-12 suggested that the two compounds were 10-epimers. The suggestion was supported by the ECD spectra of 2 and 3, showing the opposite Cotton effects at 317 nm (Δε -4.70 for 2) and 320 nm (Δε +6.36 for 3) (Fig. 4). Fortunately, 2 was successfully crystallized from acetone by the ENaCt method. Single crystal X-ray diffraction using Cu Kα radiation proved the structure of 2, having the 10S, 2′R configuration (Fig. 3) (Flack parameter [0.15(9)], and Hooft parameter [-0.18(8)]). In addition, the theoretical ECD spectra of 2 and 3 were calculated at the (B3LYP/6-311g**) level, and the predicted ECD curve of (10S, 2′R)-2 and (10R, 2′R)-3 were in good agreement with the experimental ones (Fig. 4). Therefore, the structures of 2 and 3 were established as (10S, 2′R)-2 and (10R, 2′R)-3, and called amphichoterpenoids B and C.
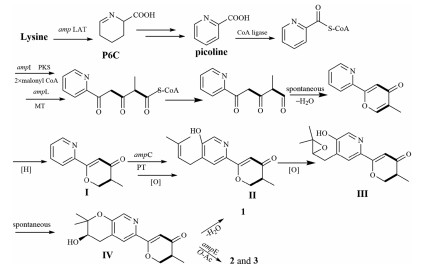
Structurally, amphichoterpenoids A–C were the first example of picoline-derived meroterpenoids with a pyrano[3, 2-c]pyridinyl-γ-pyranone skeleton, possibly deriving from lysine-terpenoid hybrid biosynthetic pathway. Here, a possible biosynthetic pathway for 1-3 was proposed (Scheme 1). Briefly, the precursor lysine (Lys) underwent aminotransfer by aminotransferase (Lys aminotransferase, LAT), spontaneously cyclized, and aromatized by oxidoreductase to furnish the picoline as the nonterpenoid starter unit [17-20]. Afterward, a picoline-derived CoA was condensed with two molecules of malonyl-CoA and an S-adenosylmethionine (SAM)-dependent methyltransferase to form the intermediate I with γ-pyranone moiety through a polyketide pathway. Then, isoprenylation of I, followed by oxidation, cyclization and reoxidation, would afford the precursor IV as the basic core structure, which undergoes either dehydration to produce (±)-1, or O-acetylation to give 2 and 3. Besides, the antiSMASH 5.2.0 and 2ndFind analysis of the related gene cluster (amp) supports the hypothetic biosynthetic pathway (Table S6 in Supporting information), despite needing to be further proved by biosynthesis. Notably, the putative Lys aminotransferase-like encoded gene (amp LAT) was located outside the amp biosynthetic gene cluster (BGC) in A. felina, and the phenomenon was also observed in the aminotransferase encoded genes in the fungus Metarhizium robertsii [18].

|
Download:
|
| Scheme 1. Proposed biosynthetic pathway for 1–3. PKS=polyketide synthase; MT=SAM dependent methyltransferase; PT=prenyltransferase; O-Ac = O-acetyltransferase. | |
{kind=link}
All of the isolates were screened for inhibitory activities against acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), which are promising therapeutic targets in Alzheimer's disease (AD) [21-24]. The compounds (+)-1, (-)-1, (±)-1, 2 and 3 displayed selective inhibition of cholinesterase, and exhibited moderate inhibitory activity against AChE with 50% inhibiting concentration (IC50) values of 18.8–53.2 μmol/L (the positive control rivastigmine, IC50 = 3.9 μmol/L). Meanwhile, none inhibited BChE (IC50 > 100 μmol/L) (Table S7 in Supporting information).
In conclusion, the rare picoline-derived meroterpenoids, amphichoterpenoids A–C possessing a pyrano[3, 2-c]pyridinyl-γ-pyranone scaffold, may attract worldwide attention of chemists and biologists. Further studies, such as in-depth biosynthetic mechanisms, are warranted.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Natural Science Foundation of China (No.41806155), the Guangdong MEPP Fund (No. GDOE (2019) A21), the National Key R & D Program of China (No. 2019YFC0312501), the Natural Science Foundation of Guangdong Province (No. 2018A030310304), and the Key-Area Research and Development Program of Guangdong Province (No. 2020B1111030005) for generous support.
| [1] |
R. Geris, T.J. Simpson, Nat. Prod. Rep. 26 (2009) 1063-1094. DOI:10.1039/b820413f |
| [2] |
Y. Matsuda, I. Abe, Nat. Prod. Rep. 33 (2016) 26-53. DOI:10.1039/C5NP00090D |
| [3] |
Y. Matsuda, I. Abe, Chapter 14, Comprehensive Natural Products Ⅲ: Chemistry and Biology, Vol. 1, Elsevier, 2020, pp. 445-478.
|
| [4] |
M. Jiang, Z. Wu, L. Liu, S. Chen, Org. Biomol. Chem. 19 (2021) 1644-1704. DOI:10.1039/D0OB02162H |
| [5] |
M.D. Sintchak, M.A. Fleming, O. Futer, et al., Cell 85 (1996) 921-930. DOI:10.1016/S0092-8674(00)81275-1 |
| [6] |
S. Liu, J. Widom, C.W. Kemp, C.M. Crews, J. Clardy, Science 282 (1998) 1324-1327. DOI:10.1126/science.282.5392.1324 |
| [7] |
J.M. Molina, M. Tourneur, C. Sarfati, et al., New England J. Med. Surg. Collat. Branches Sci. 346 (2002) 1963-1969. |
| [8] |
M.C. Chen, T.Y. Cho, Y.H. Kuo, T.H. Lee, J. Nat. Prod. 80 (2017) 2439-2446. DOI:10.1021/acs.jnatprod.7b00223 |
| [9] |
T. Itoh, K. Tokunaga, Y. Matsuda, et al., Nat. Chem. Biol. 2 (2010) 858-864. DOI:10.1038/nchem.764 |
| [10] |
M. Jiang, Z. Wu, H. Guo, L. Liu, S. Chen, Mar. Drugs 18 (2020) 321-368. DOI:10.3390/md18060321 |
| [11] |
A. El-Demerdash, D. Kumla, A. Kijjoa, Mar. Drugs 18 (2020) 317-348. DOI:10.3390/md18060317 |
| [12] |
S. Chen, M. Jiang, B. Chen, et al., Mar. Drugs 17 (2019) 522-531. DOI:10.3390/md17090522 |
| [13] |
S. Chen, H. Shen, P. Zhang, et al., Chem. Commun. (Camb.) 55 (2019) 1438-1441. DOI:10.1039/C8CC08970A |
| [14] |
S.I. Niaz, P. Zhang, H. Shen, et al., Nat. Prod. Res. 33 (2019) 1262-1268. DOI:10.1080/14786419.2018.1470173 |
| [15] |
S. Chen, H. Shen, Y. Deng, et al., Mar. Life Sci. Tech. 3 (2021) 69-76. DOI:10.1007/s42995-020-00066-8 |
| [16] |
A.R. Tyler, R. Ragbirsingh, C.J. McMonagle, et al., Chemistry 6 (2020) 1755-1765. DOI:10.1016/j.chempr.2020.04.009 |
| [17] |
M. Lv, J. Zhao, Z. Deng, Y. Yu, Chem. Biol. 22 (2015) 1313-1324. DOI:10.1016/j.chembiol.2015.09.005 |
| [18] |
F. Luo, S. Hong, B. Chen, et al., ACS Chem. Biol. 15 (2020) 2476-2484. DOI:10.1021/acschembio.0c00466 |
| [19] |
T. Fujii, M. Mukaihara, H. Agematu, H. Tsunekawa, Biosci. Biotechnol. Biochem. 66 (2002) 622-627. DOI:10.1271/bbb.66.622 |
| [20] |
G.J. Gatto Jr., M.T. Boyne 2nd, N.L. Kelleher, C.T. Walsh, J. Am. Chem. Soc. 128 (2006) 3838-3847. DOI:10.1021/ja0587603 |
| [21] |
H. Li, W. Feng, X. Li, et al., Org. Lett. 22 (2020) 7041-7046. DOI:10.1021/acs.orglett.0c02641 |
| [22] |
S. Darvesh, D.A. Hopkins, C. Geula, Nat. Rev. Neurosci. 4 (2003) 131-138. DOI:10.1038/nrn1035 |
| [23] |
A.G. Zaki, E.R. El-Sayed, M. Abd Elkodous, G.S. El-Sayyad, Appl. Microbiol. Biot. 104 (2020) 4717-4735. DOI:10.1007/s00253-020-10560-9 |
| [24] |
P.J. Houghton, Y. Ren, M.J. Howes, Nat. Prod. Rep. 23 (2006) 181-199. DOI:10.1039/b508966m |