 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Functional Small Organic Molecules, Ministry of Education, and College of Chemistry & Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China;
c State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China;
d School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
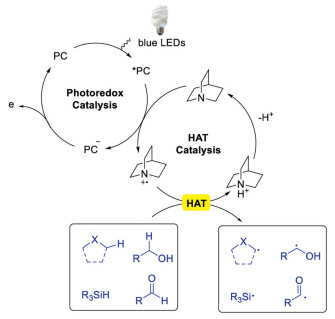
The direct functionalization of C-H bonds is one of the most attractive protocols to construct molecules in organic chemistry as it skips the steps of pre-activation, promoting reaction in an atom- and step-economy way [1-10]. Thus, continuous efforts have been made in developing direct C-H functionalization strategies. Recently, the direct C(sp3)-H functionalization through visible light mediated hydrogen-atom-transfer (HAT) has attracted great interests [10-13]. HAT, a concerted movement of a proton and an electron between two compounds in a single step, allows the direct and selective functionalization of C-H bonds. Accordingly, the reactivity of HAT can be tuned by HAT catalyst. Consequently, a variety of HAT catalysts, such as thiols [14-16] and benzophenones [17-20] have been developed to activate substrates with various functional groups. The reactivity of these HAT catalysts largely depends on the bond-dissociation-enthalpy (BDE) of the substrates. Therefore, it is unable to abstract the hydrogen of strong C-H bonds, which limits the utilization of this powerful method. However, the reactivity of C-H bond abstraction depends not only on BDE, but also on polar effects of the transition state. Quinuclidine, a novel HAT catalyst disclosed by Macmillan's group in 2015 [21], exhibited complementary reactivity toward other HAT catalysts. Mechanistic studies indicated that quinuclidine (E1/2ox = +1.1 V vs. SCE in MeCN) was able to quench the photoexcited catalyst, such as Ir[dF(CF3)ppy]2(dtbbpy)PF6 (E1/2red = +1.21 V vs. SCE in MeCN), to generate quinuclidine radical cation. The quinuclidine radical cation was more electrophilic, and the BDE of protonated quinuclidium ion (H-N+ BDE =100 kcal/mol) was higher than other HAT catalysts. These features enabled the hydrogen abstraction of stronger and hydric C-H bonds by quinucindine radical cation and its derivatives in the presence of weaker and neutral C-H bonds (Scheme 1).

|
Download:
|
| Scheme 1. Activation pattern of quinuclidine as HAT catalyst under visible light irradiation. | |
{kind=link}
This review summarizes the recent advance of visible light mediated reactions with quinuclidine and its derivatives as the HAT catalysts. The manuscript is classified by the type of substrates, for instance, alcohols, amines and ethers, aldehydes, R3SiH, adamantanes, carboxylates and DMF.
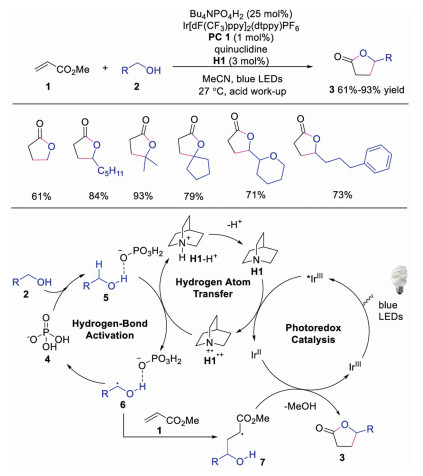
2. Hydrogen atom abstraction of alcoholHydroxyl group is widely existed in natural products. However, direct α-H functionalization of hydroxyl group is quite difficult due to the strong C-H bonds. It was demonstrated that intermolecular hydrogen bonding between hydrogen bonding acceptors and alcohols would lead to polarization and weakening of adjacent C-H bonds. Based on this strategy, Macmillan's group reported the direct C-H functionalization at the α-H of hydroxyl group by using quinuclidine as the HAT catalyst and tetra-n-butylammonium phosphate (TBAP) as the hydrogen bond acceptor in 2015 [21]. A variety of lactones 3, generated by alcohols 2 and activated alkenes 1, could be efficiently produced by the triple catalysis of photoredox, HAT and hydrogen-bond activation. The electron-deficient quinuclidine radical cation, could selectively abstract α-H of alcohol under the activation of TBAP. This was the first example using quinuclidine as the HAT catalyst. The authors proposed a mechanism in Scheme 2. The photoexcited *IrⅢ catalyst was quenched by quinuclidine to afford IrⅡ along with quinuclidine radical cation H1·+. In the meantime, the interaction between TBAP and hydroxyl group of alcohol would make the α-H more hydridic, which was abstracted by quinuclidine radical cation H1·+ to form α-hydroxyl radical 6 and quinuclidine ion. Further deprotonation of quinuclidine ion afforded quinuclidine to complete the HAT cycle. On the other hand, the interaction between α-hydroxyl radical 6 and activated alkenes 1 provided a radical intermediate 7, which was reduced by IrⅡ. The subsequent lactonization would furnish the final product 3. This triple catalysis system could directly activate the α-H of hydroxyl group, providing an efficient strategy for direct α-functionalization of alcohols.

|
Download:
|
| Scheme 2. C-Alkylation of alcohols under triple catalysis of photoredox, HAT and TBAP. | |
{kind=link}
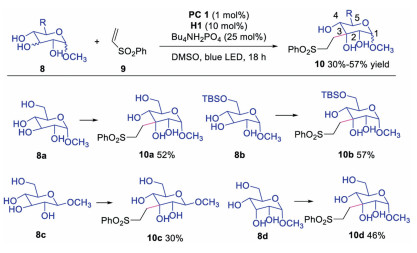
Later, Minnaard and co-workers applied this triple catalysis to access functionalized carbohydrates with excellent regioselectivity from unprotected glucosides, xyllosides and allosides in 2017 (Scheme 3) [22]. The reaction showed excellent regioselectivity for secondly hydroxyl group at C3 in the presence of various hydroxyl groups. The primary hydroxyl group protected substrate 8b provided product 10b in slightly better yield. It is interesting to note that substrate 8c with equatorial C1-OMe and 8d with C3-axial hydroxyl group also afforded C3-equatorial functionalized products 10c and 10d. However, mechanism for the regioselectivity was not clear and deserved for further studies.

|
Download:
|
| Scheme 3. Functionalization of carbohydrates under catalysis of photoredox, HAT and TBAP. | |
{kind=link}
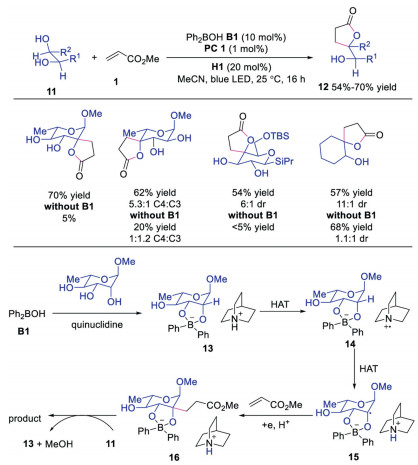
In 2019, Taylor and co-workers further developed the combination of photoredox catalyst, quinuclidine and diphenylborinic acid to realize stereo- and siteselective C-H alkylation of carbohydrates derivatives [23]. Diverse cis-diols compounds could be selectively functionalized at the α-carbon of alcohol with activated alkene, affording the corresponding products with retention of configuration. The results showed significant improvement in reactivity, site- and regioselectivity with diphenylborinic acids, indicating that diphenylborinic acid played an important role in this transformation. A plausible mechanism was proposed as shown in Scheme 4. Initially, diphenylborinic acid B1 interacted with diol 11 to furnish tetracoordinate borinic ester 13, which underwent HAT with quinuclidine radical cation to provide intermediate 14. This intermediate 14 would accelerate the HAT between tetracoordinate diarylborinic ester and quinuclidine radical cation due to ion pairing or polarity matching effects to afford intermediate 15. Subsequently, radical anion 15 reacted with activated alkene 1, followed by SET and protonation to provide intermediate 16. After deboronation, the final product would be produced. This strategy extended the utility of the above triple catalysis system to generate functionalized carbohydrates from cis-diols.

|
Download:
|
| Scheme 4. Functionalization of carbohydrates under triple catalysis of photoredox, HAT and diphenylborinic acid. | |
{kind=link}
Taylor's group reported the synthesis of 2-keto-3-deoxyfuranosides through triple catalysis system of photoredox, HAT and boron in 2020 [24]. A range of furanoside derivatives, which were challenging to be obtained through other methods, were efficiently produced in moderate to good yield with good C2 regioselectivity. Mechanism studies showed that boronic ester, generated by cis-1, 2-diol group and organoboron catalyst, was able to facilitate the abstraction of hydrogen to form α-hydroxyl radical. The mechanism was presented in Scheme 5. The diol would react with boron and quinuclidine to afford intermediate 21, which underwent HAT with quinuclidine radical cation to generate α-hydroxyl radical 22. Subsequently, C3-O bond cleavage of α-hydroxyl radical 22 provided α-keto radical 23. Followed by SET and protonation, the final product would be produced. This method provided an efficient approach to access precursors to modified carbohydrate and nucleoside derivatives.

|
Download:
|
| Scheme 5. Synthesis of furanoside derivatives under photoredox, HAT and boron catalysis. | |
{kind=link}
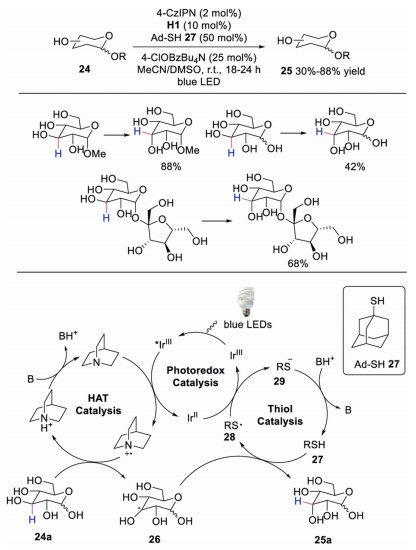
Recently, Wendlandt and co-workers disclosed an efficient way to prepare rare sugar isomers via site-selective epimerization from biomass carbohydrates [25]. The reaction was promoted by photoredox catalyst and two distinct HAT catalysts. A variety of biomass-derived and abundant monosaccharides were well tolerated, affording the corresponding rare sugars in moderate to good yields. The epimerization proceeded through hydrogen atom abstraction and hydrogen atom donation with quinuclidine and thiols. A proposed mechanism was shown in Scheme 6. It was reasoned that the photoexcited *IrⅢ was quenched by quinuclidine to generate quinclidine radical cation, which subsequently underwent HAT with compound 24a affording α-hydroxyl radical intermediate 26. Mechanistic studies indicated that thiol radical 28 was not able to promote the hydrogen atom transfer process. Therefore, the thiol acidity was a key parameter as the photoexcited *IrⅢ could only be quenched by quinuclidine radical cation instead of thiol anion. Next, α-hydroxyl radical abstracted a hydrogen from thiol 27 leading to the final product 25a. The epimerization process was a kinetically controlled process. This reaction provided a simple and potentially extensive approach to synthesize a valuable class of natural products.

|
Download:
|
| Scheme 6. Synthesis of rare sugars from biomass carbohydrates. | |
{kind=link}
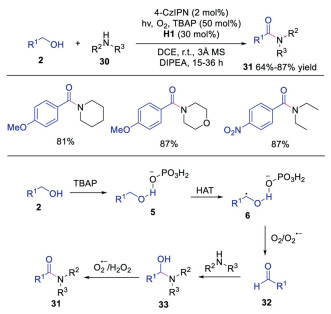
In the same year, Singh's group extended the triple catalysis of photoredox, HAT and hydrogen bonding catalysis to the synthesis of amides from alcohols and amines under aerobic conditions [26]. A wide range of alcohols and amines were suited for this reaction, giving rise to the corresponding products in good yields. As shown in Scheme 7, α-hydroxyl radical 6, generated from intermediate 5 via HAT with quinuclidine radical cation, was oxidized to aldehyde 32. Subsequently, this aldehyde 32 underwent condensation with amine to afford hemiaminal 33, which was oxidized to produce the final amide product. This triple catalysis system provided a concise method for the synthesis of amides with various functional groups.

|
Download:
|
| Scheme 7. Synthesis of amides under triple catalysis of photoredox, HAT and TBAP. | |
{kind=link}
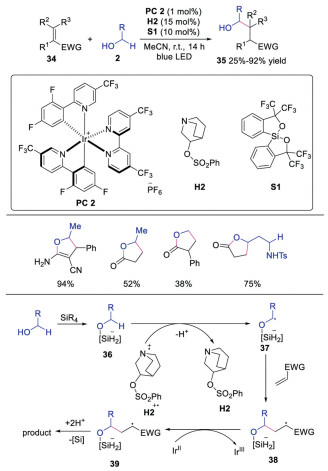
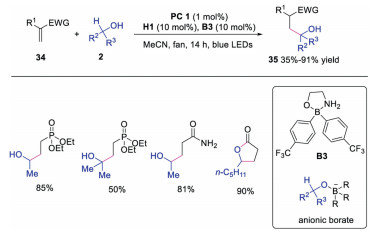
In 2020, Kanai's group developed α-functionalization reaction of alcohols with photoredox, HAT and a novel bond-weakening catalyst, Martin's spirosilane [27]. A wide range of activated alkenes and alcohols were suitable in the reaction, affording the corresponding products in moderate to good yields. The mechanism shown in Scheme 8 revealed that alcohol 2 would react with Martin's spirosilane S1 leading to pentavalent anionic silicate species 36. DFT calculations indicated that the anionic silicate species 36 could efficiently weaken the BDE of α-C–H. Subsequently, HAT between anionic silicate species 36 and radical cation H2·+ provided α-hydroxyl radical 37, which reacted with activated alkene giving rise to radical intermediate 38. This intermediate 38 would be reduced by IrⅡ species. Further protonation or lactonization produced the final product. The novel bond-weakening catalyst might provide potential for exploring unprecedented selectivity, reactivity and substrate generality in direct C-H functionalization reactions. Later, the same group also discovered that borinic acid-ethanolamine could perform as the bond weakening catalyst (Scheme 9) [28]. The in situ generated anionic borates would weaken the BDE of α-C-H. This bond weakening reagent could broaden substrate scope and enhance reactivity, probably due to the stronger bond-weakening effect.

|
Download:
|
| Scheme 8. C-Alkylation of alcohols under triple catalysis of photoredox, HAT and Martin's spirosilane. | |
{kind=link}

|
Download:
|
| Scheme 9. C-Alkylation of alcohols under triple catalysis of photoredox, HAT and borinic acid-ethanolamine. | |
{kind=link}
Yang and co-workers reported the synthesis of 1, 4-carbonyl compounds through isomerization of γ-carbonyl allylic alcohols under triple catalysis of photoredox, HAT and hydrogen-bonding catalyst [29]. A range of allylic alcohols were employed in this isomerization reaction, even those bearing tetra-substituted olefins. A plausible mechanism was proposed, as shown in Scheme 10. Intermediate 42 from γ-carbonyl allylic alcohol 40 and TBAP underwent HAT with quinuclidine radical cation leading to α-hydroxyl radical 43. DFT calculations indicated that α-hydroxyl radical 43 with γ-electron-deficient substitutions would facilitate the single electron transfer (SET) process with reduced photocatalyst, affording allylic anion 44. This anion would subsequently undergo protonation and tautomerization to provide 1, 4-dicarbonyl compound 41. The approach provided a mild way to generate 1, 4-dicarbonyl compounds via isomerization of allylic alcohols.

|
Download:
|
| Scheme 10. Isomerization of highly substituted allylic alcohols. | |
{kind=link}
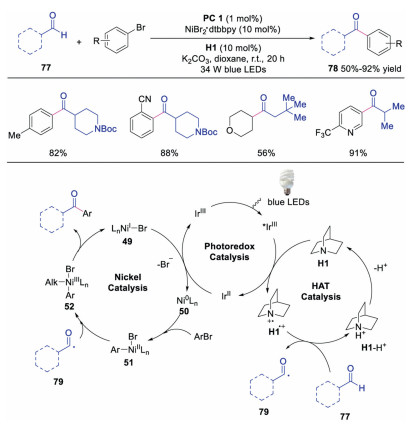
Macmillan and co-workers further developed the triple nickel, HAT and photoredox catalysis for the direct α-arylation of alcohols [30]. The key step of this transformation was the interaction of Lewis acid with alcohol affording a metal alkoxide species, which prevented the undesired oxygen transmetallation with NiⅡaryl catalytic intermediate. Benzylic alcohols with different functional groups, which were important motifs in drug discovery, could be efficiently obtained. The authors proposed a mechanism as shown in Scheme 11. The photoexcited *IrⅢ would be quenched by quinuclidine to afford quinuclidine radical cation along with a IrⅡ species. Meanwhile, the interaction between Lewis acid and alcohol furnished Lewis acid-coordinated alkoxide 47, which underwent HAT with quinuclidine radical cation to produce α-alkoxyl radical 48. Subsequently, NiⅡ intermediate 51, formed from the oxidative addition of Ni0 species 50 to aryl halide 45, would react with α-alkoxyl radical 48 to generate NiⅢ species 52. Finally, reductive elimination of NiⅢ species 52 gave rise to the final product and NiI species 49, which would go through SET with IrⅡ to regenerate Ni0 catalyst 50 and photocatalyst.

|
Download:
|
| Scheme 11. Synthesis of benzylic alcohols under triple catalysis of photoredox, HAT and nickel salt. | |
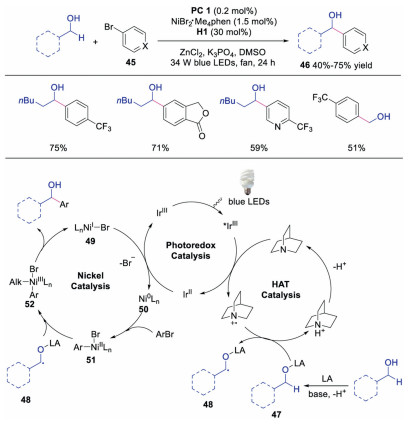{kind=link}
3. Hydrogen atom abstraction of amine, ethers and thioethers
In 2016, Macmillan and co-workers described the direct functionalization of α-amino and α-oxy C–H activation with aryl halides under triple catalysis of photoredox, HAT and nickel catalyst [31]. The radical cation from quinuclidine derivative, was able to selectively abstract the electron-rich, hydridic C-H bond. A wide array of amines and ethers could participate in the reaction well, affording the corresponding arylation products in moderate to good yields. The proposed mechanism (Scheme 12) showed that amine would undergo HAT with quinuclidine radical cation to generate α-amino radial 56. Meanwhile, oxidative addition between Ni0 catalyst 50 and aryl halide would occur leading to NiⅡ species 51, which would react with α-amino radial 56 to provide NiⅢ species 52. The final product would be afforded upon reductive elimination of NiⅢ species 52. This protocol provided a simple and efficient way by using compounds possessing C-H bonds as organometallic nucleophile equivalents. In 2017, the same group extended the triple catalysis of nickel, photoredox and HAT to the direct C-H alkylation reactions at α site of amines and ethers with alkyl halides [32], and the corresponding alkylation derivatives were formed in moderate to good yields with good to excellent regioselectivity (Scheme 13). This method might provide a novel approach to late-stage functionalization.

|
Download:
|
| Scheme 12. Arylation of amines and ethers under triple catalysis of photoredox, HAT and nickel salt. | |
{kind=link}

|
Download:
|
| Scheme 13. Alkylation of amines and ethers under triple catalysis of photoredox, HAT and nickel salt. | |
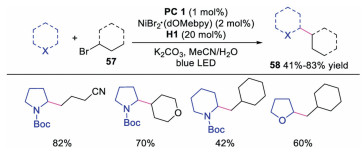{kind=link}
Direct α-functionalization of primary amines is a significant challenge, due to their higher oxidation potential and inherent nucleophilicity and basicity. In 2018, Rovis' group successfully realized the direct α-functionalization of primary amines with CO2 under visible light irradiation and HAT catalysis [33]. Diverse γ-lactams were provided in moderate to good yields. The authors proposed a plausible mechanism, as shown in Scheme 14. Carbamates 62 generated in situ from primary amine 59 and CO2 would be functionalized at less reactive C-H bonds, since the formation of alkylammonium carbamate would dramatically diminish the nucleophilicity of amino group. Therefore, the carbamate would go through HAT with quinuclidine radical cation to provide radical intermediate 63, which subsequently reacted with activated alkene 60 affording radical 64. Further SET and lactamization of radical 64 would produce the final product. This protocol provided a concise way to direct functionalization of primary amines at the α-site without protection and deprotection steps.

|
Download:
|
| Scheme 14. α-Functionalization of primary amines. | |
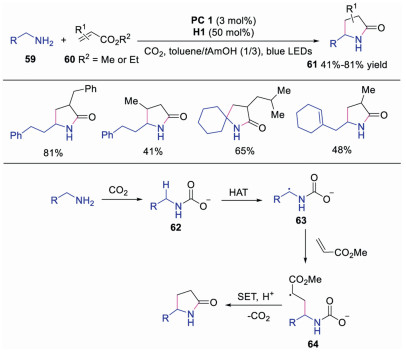{kind=link}
The site-selective α-functionalization of amines could be further extended [34]. The same group described the generation of α-functionalized amine derivatives in moderate to good yield with excellent regioselectivity. The use of trifluoromethanesulfonyl group on primary amine was the key to access the α-selectivity. It was reasoned that various protecting groups on primary amine would result in different C-H bond strength and pKa value of NH, thus delivering significant divergence reactivity. Computational studies revealed that nitrogen radical from triflamide was more stable than that from trifluoroacetamide. Therefore, the trifluoroacetamide radical would undergo intermolecular [1, 5] HAT due to the instability of nitrogen radical, while the more stable nitrogen radical underwent HAT with quinuclidine radical cation. On the basis of control experiments and mechanism studies, a plausible mechanism was proposed as shown in Scheme 15. The trifluoromethanesulfonyl protected amine was deprotonated in the presence of base leading to nitrogen anion 68, which go through HAT with quinuclidine radical cation to produce α-amine radical 69. Subsequently, α-amine radical 69 reacted with activated alkene to generate radical intermediate 70, which underwent SET and protonation giving rise to the final product. In this transformation, quinuclidine acted as both base and HAT catalyst. This methodology provided a general way for α-functionalization of primary amine derivatives.

|
Download:
|
| Scheme 15. α-Functionalization of primary amine derivatives. | |
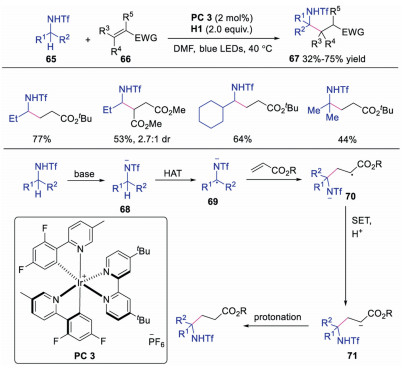{kind=link}
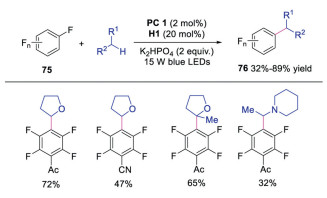
In 2019, Wang and co-workers described the inert C(sp3)-H monofluoroalkenylation of heteroalkanes with difluoroethylene via dual photoredox and quinuclidine HAT catalysis (Scheme 16) [35]. The corresponding monofluoroalkenylation products were afforded in moderate to good yields. Mechanistic studies showed that the photoexcited IrⅢ was quenched by quinuclidine to form quinuclidine radical cation, which abstracted an inert hydrogen of heteroalkanes to produce radical intermediate 56. In the meantime, diphenyl gem-difluoroethylene was reduced by IrⅡ to furnish fluoroalkenyl radical 74. The radical coupling between radical 56 and radical 74 would afford the final product. This reaction was an advance in monofluoroalkenylation under mild conditions with high regioselectivity and broad substrate scope. In the same year, Xia's group reported the visible light driven C(sp3)-C(sp2) coupling between perfluoarenes and ethers by using similar methodology with quinuclidine as the HAT catalyst (Scheme 17) [36].

|
Download:
|
| Scheme 16. Monofluoroalkenylation of heteroalkanes with difluoroethylene. | |
{kind=link}

|
Download:
|
| Scheme 17. C-H Multifluoroarylation reaction. | |
{kind=link}
4. Hydrogen atom abstraction of aldehyde
Ketone moieties are widely existed in natural products and pharmaceuticals. Moreover, they have been utilized broadly in organic synthesis. Therefore, generation of functionalized ketones has attracted great interests. In 2017, Macmillan's group reported the synthesis of ketones through direct C–H functionalization of aldehydes under triple catalysis of HAT, photoredox and Ni salt (Scheme 18) [37]. The corresponding ketones could be obtained in moderate to good yields with excellent formyl C-H selectivity. Mechanistic studies showed that the formyl hydrogen of aldehyde was abstracted selectively by quinuclidine radical cation to form acyl radical 79. Subsequently, this acyl radical 79 reacted with NiⅡ species 51 affording NiⅢ species 52. After reductive elimination, ketone would be produced. In 2020, Iaroshenko's group used the same triple catalysis to synthesize 3-acyl substituted chromones (Scheme 19) [38]. The 3-acyl substituted chromones could be obtained either by pathway a (3-halogenchromones and aldehyde) or pathway b (3-formylchromones and aryl halide). Diverse chromones were formed in an atom-economic manner in good to excellent yields.

|
Download:
|
| Scheme 18. Direct C-H functionalization of aldehyde. | |
{kind=link}

|
Download:
|
| Scheme 19. Synthesis of 3-acyl substituted chromones. | |
{kind=link}
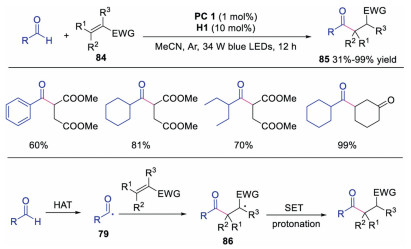
In 2017, Liu and co-workers described the C(sp2)-H functionalization of aldehydes with activated alkenes through dual photoredox and HAT catalysis (Scheme 20) [39]. The acyl radical 79 could be directly formed with quinuclidine as the HAT catalyst without pre-functionalization and transition metal catalysis. A wide range of unsymmetric ketones could be afforded in moderate to good yields. The acyl radical 79, generated from aldehyde via HAT, would attack the activated alkene leading to radical 86. Further SET and protonation would produce the ketone product.

|
Download:
|
| Scheme 20. C(sp2)-H Functionalization of aldehydes. | |
{kind=link}
5. Hydrogen atom abstraction of R3SiH compounds
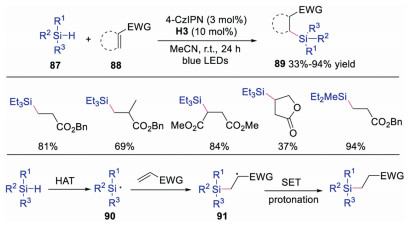
In 2017, Wu and co-workers developed a metal-free hydrosilylation of alkenes via dual photoredox and HAT catalysis using quinuclidine-derivatives (Scheme 21) [40]. Compared with this method, previous works exhibited poor selectivity and reactivity. Diverse silicon-containing compounds were obtained in an atom and redox economic way with good to excellent yields. Mechanistic studies showed that silyl radical 90 was formed via HAT with quinuclidine radical cation, since the Si-H bond were more hydridic than C-H bond. Subsequently, silyl radical 90 would attack electron-deficient alkene giving rise to radical intermediate 91, which underwent SET and protonation to afford the final product. This dual catalytic system provided a practical way for the hydrosilylation of electron-deficient alkenes.

|
Download:
|
| Scheme 21. Hydrosilylation of electron-deficient alkenes. | |
{kind=link}
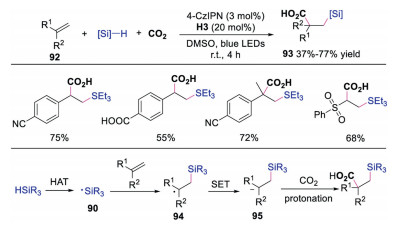
Later, the same group reported the synthesis of β-silacarboxylic acids via difunctionalization of electron-deficient alkenes using silanes and CO2 through dual photoredox and HAT catalysis [41]. The proposed mechanism shown in Scheme 22 revealed that silane underwent HAT to produce silyl radical 90, which would react with electron-deficient alkene affording radical intermediate 94. This radical 94 was further reduced to anion 95, which went through carboxylation with CO2 and protonation to generate β-silacarboxylic acid. This study was a rare example of catalytic difunctionalizaiton of activated alkenes through Si-H activation in a redox-neutral manner than an oxidative fashion.

|
Download:
|
| Scheme 22. Synthesis of β-silacarboxylic acids via difunctionalization of electron-deficient alkenes. | |
{kind=link}
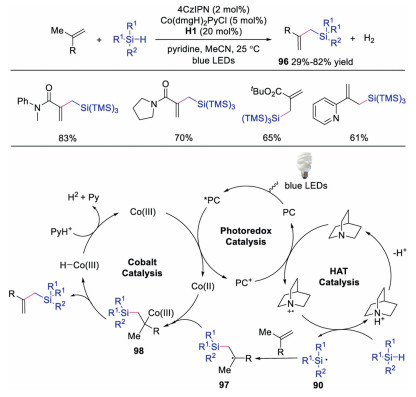
In 2019, Xu and co-workers described the synthesis of allylic silanes through dehydrogenative silylation of alkenes via triple catalysis of HAT, photoredox and cobalt salt (Scheme 23) [42]. The cobalt catalysis could avoid the hydrosilylation process and selectively afford the allylic silane products. In contrast to Wu's work, the photoexcited photocatalyst was quenched by CoⅡ species to produce CoⅢ species. In the meantime, the silicon compound underwent HAT with quinuclidine radical cation to form silyl radical intermediate 90, which reacted with activated alkene to afford radical 97. Next, the radical 97 was captured by CoⅢ species leading to intermediate 98. Then, β-hydride elimination would occur, affording allylic silane along with CoⅢ-H species. This CoⅢ-H species would further release H2 to regenerate the cobalt catalyst. This oxidant free system provided a more efficient and cleaner protocol for the synthesis of allylic silanes.

|
Download:
|
| Scheme 23. Synthesis of allylic silanes. | |
{kind=link}
6. Hydrogen atom abstraction of adamantanes
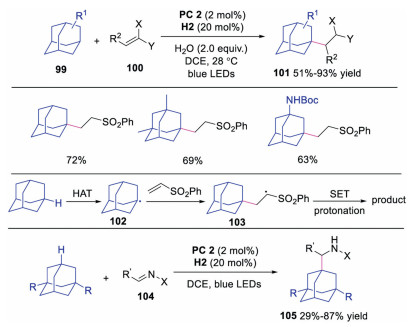
In 2019, Martin and co-workers developed the direct C–H functionalization of adamantanes via dual photoredox and HAT catalysis [43]. A novel quinuclidine derivative was utilized as the HAT catalyst to promote the tertiary C-H abstraction of adamantanes. A variety of functionalized adamantanes were obtained in good to excellent yields. The proposed mechanism was shown in Scheme 24. It was reasoned that the adamantane underwent HAT to afford adamantly radical 102, which subsequently attacked the activated alkene giving rise to radical intermediate 103. Further SET and protonation would produce the functionalized adamantane. Later, the same group also disclosed the direct C-H aminoalkylation reaction using similar strategy [44].

|
Download:
|
| Scheme 24. Direct C-H functionalization of adamantanes. | |
{kind=link}
7. Hydrogen atom abstraction of carboxylate
In 2020, Li and co-workers developed a carboarylation reaction of styrenes with aryl halide and CO2 via dual catalysis of photoredox and HAT catalysis [45]. Alkyl halides, pyridyl halides, even aryl chlorides were well tolerated, affording the corresponding hydrocinnamic acid derivatives in moderate to good yields. The proposed mechanism in Scheme 25 showed that the photoexcited catalyst was quenched by DABCO to afford DABCO·+, which underwent HAT with HCO2K to produce CO2 radical anion. Subsequently, aryl halide was reduced by CO2 radical anion to generate aryl radical 109, which attacked alkene to provide radical intermediate 110. Radical 110 was further reduced by IrⅡ leading to carbon anion 111, which captured CO2 to provide carboxylate 112. Subsequently, methylation of carboxylate 112 afforded the final product. This method offered a new protocol for Meerwein arylation-addition reactions under visible light conditions by using aryl halides as the radical sources.

|
Download:
|
| Scheme 25. Carboarylation reaction of styrenes. | |
{kind=link}
8. Hydrogen atom abstraction of DMF
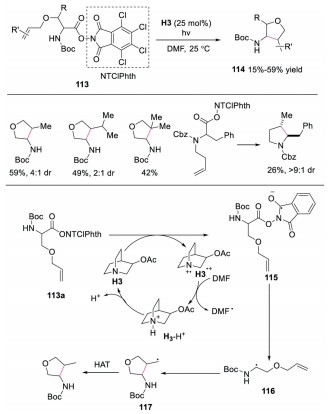
In 2019, Bach and co-workers reported the synthesis of functionalized furans and pyrroles by using quinuclidine derivative as the electron-donor catalyst to promote intramolecular cyclization via EDA complex under visible light conditions [46]. The authors proposed a plausible mechanism as shown in Scheme 26. The EDA complex, formed by 113a and quinuclidine derivative H3, underwent SET to generate H3·+ and radical intermediate 115. Subsequently, decarboxylation of 115 would occur, providing radical intermediate 116. This radical intermediate 116 would further undergo intramolecular cyclization to generate radical 117, which went through HAT to produce the final product. This was the first example in which a catalytic amount of quinuclidine derivative acted as the electron donor in EDA complex.

|
Download:
|
| Scheme 26. Photoredox and HAT catalyzed intramolecular cyclization reaction. | |
{kind=link}
9. Summary and outlook
In summary, quinuclidine and its derivatives can act as HAT catalyst under visible light irradiation. Compared with other type of HAT catalysts, such as thiols and benzophenones, it tends to abstract the hydrogen from electron-rich or hydridic C-H bonds in the presence of weak and neutral C-H bonds. With hydrogen bond acceptors, alcohols can be selectively functionalized at the α site of hydroxyl group with quinuclidine under visible light irradiation. Furthermore, direct C-H functionalization of amines, ethers, thioethers, aldehydes, SiH compounds, adamantanes and carboxylates can efficiently take place as well by using quinuclidine and its derivatives as the HAT catalysts. Additionally, quinuclidine derivatives can act as the electron donor to catalyze reactions through EDA complexes with electron acceptors. The reported results show that quinuclidine and its derivatives exhibit great catalytic ability toward the activation of various compounds, and these methods provide an alternative way for the generation of bioactive and natural compounds.
Despite the significant achievements of quinuclidine and its derivatives as HAT catalysts, there are still many issues to be solved. Quinuclidine has shown great power for the synthesis of racemic compounds, while asymmetric reactions are less studied. Therefore, quinuclidine as HAT catalyst may be employed with chiral catalytic system to produce chiral compounds. Meanwhile, quniuclidine and its derivatives can serve as Lewis base to activate unsaturated compounds such as alkenes and allenes, and the combination of two characters of quinuclidine may provide new avenues for quinuclidine-catalyzed reactions. We believe that quinuclidine as HAT catalyst is promising for further development.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsFinancial support from National Natural Science Foundation of China (Nos. 21871053 and 21532001), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2020ZD04) is gratefully acknowledged.
| [1] |
E.V. Verbitskiy, G.L. Rusinov, O.N. Chupakhin, V.N. Charushin, Synthesis 50 (2018) 193-210. DOI:10.1055/s-0036-1589520 |
| [2] |
M. Monika, S. Selvakumar, Synthesis 51 (2019) 4113-4136. DOI:10.1055/s-0037-1611910 |
| [3] |
A.C. Sun, R.C. McAtee, E.J. McClain, C.R.J. Stephenson, Synthesis 51 (2019) 1063-1072. DOI:10.1055/s-0037-1611658 |
| [4] |
A. Biswas, S. Maity, S. Pan, R. Samanta, Chem. Asian J. 15 (2020) 2092-2109. DOI:10.1002/asia.202000506 |
| [5] |
E. Kang, H.T. Kim, J.M. Joo, Org. Biomol. Chem. 18 (2020) 6192-6210. DOI:10.1039/D0OB01265C |
| [6] |
G. Kuang, G. Liu, X. Zhang, et al., Synthesis 52 (2020) 993-1006. DOI:10.1055/s-0039-1690816 |
| [7] |
Y. Li, X.F. Wu, Angew. Chem. Int. Ed. 59 (2020) 1770-1774. DOI:10.1002/anie.201914914 |
| [8] |
S. Liu, G.J. Deng, H. Huang, Synlett 32 (2021) 142-158. DOI:10.1055/s-0040-1707217 |
| [9] |
S. Yakubov, J.P. Barham, Beilstein J. Org. Chem. 16 (2020) 2151-2192. DOI:10.3762/bjoc.16.183 |
| [10] |
H. Yorimitsu, A. Yoshimura, Y. Misaki, Synthesis 52 (2020) 3326-3336. DOI:10.1055/s-0040-1707256 |
| [11] |
S. Protti, M. Fagnoni, D. Ravelli, ChemCatChem 7 (2015) 1516-1523. DOI:10.1002/cctc.201500125 |
| [12] |
L. Capaldo, D. Ravelli, Eur. J. Org. Chem. 2017 (2017) 2056-2071. DOI:10.1002/ejoc.201601485 |
| [13] |
L. Capaldo, L.L. Quadri, D. Ravelli, Green Chem. 22 (2020) 3376-3396. DOI:10.1039/D0GC01035A |
| [14] |
D. Hager, D.W.C. MacMillan, J. Am. Chem. Soc. 136 (2014) 16986-16989. DOI:10.1021/ja5102695 |
| [15] |
K. Qvortrup, D.A. Rankic, D.W.C. MacMillan, J. Am. Chem. Soc. 136 (2014) 626-629. DOI:10.1021/ja411596q |
| [16] |
J.D. Cuthbertson, D.W.C. MacMillan, Nature 519 (2015) 74-77. DOI:10.1038/nature14255 |
| [17] |
S. Kamijo, G. Takao, K. Kamijo, et al., Angew. Chem. Int. Ed. 55 (2016) 9695-9699. DOI:10.1002/anie.201603810 |
| [18] |
S. Kamijo, G. Takao, K. Kamijo, et al., Org. Lett. 18 (2016) 4912-4915. DOI:10.1021/acs.orglett.6b02391 |
| [19] |
Y. Amaoka, M. Nagatomo, M. Watanabe, et al., Chem. Sci. 5 (2014) 4339-4345. DOI:10.1039/C4SC01631A |
| [20] |
N. Ishida, Y. Masuda, S. Uemoto, M. Murakami, Chem. Eur. J. 22 (2016) 6524-6527. DOI:10.1002/chem.201600682 |
| [21] |
J.L. Jeffrey, J.A. Terrett, D.W.C. MacMillan, Science 349 (2015) 1532-1536. DOI:10.1126/science.aac8555 |
| [22] |
I.C. Wan, M.D. Witte, A.J. Minnaard, Chem. Commun. (Camb.) 53 (2017) 4926-4929. DOI:10.1039/C7CC01416C |
| [23] |
V. Dimakos, H.Y. Su, G.E. Garrett, M.S. Taylor, J. Am. Chem. Soc. 141 (2019) 5149-5153. DOI:10.1021/jacs.9b01531 |
| [24] |
V. Dimakos, D. Gorelik, H.Y. Su, et al., Chem. Sci. 11 (2020) 1531-1537. DOI:10.1039/C9SC05173B |
| [25] |
Y. Wang, H.M. Carder, A.E. Wendlandt, Nature (2020) 403-408. |
| [26] |
S.S. Shah, M. Shee, Y. Venkatesh, et al., Chem. Eur. J. 26 (2020) 3703-3708. DOI:10.1002/chem.201904924 |
| [27] |
K. Sakai, K. Oisaki, M. Kanai, Adv. Synth. Catal. 362 (2020) 337-343. DOI:10.1002/adsc.201901253 |
| [28] |
K. Sakai, K. Oisaki, M. Kanai, Synthesis-Stuttgart 52 (2020) 2171-2184. DOI:10.1055/s-0040-1707114 |
| [29] |
K. Guo, Z. Zhang, A. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 11660-11668. DOI:10.1002/anie.202000743 |
| [30] |
J. Twilton, M. Christensen, D.A. DiRocco, et al., Angew. Chem. Int. Ed. 57 (2018) 5369-5373. DOI:10.1002/anie.201800749 |
| [31] |
M.H. Shaw, V.W. Shurtleff, J.A. Terrett, J.D. Cuthbertson, D.W.C. MacMillan, Science 352 (2016) 1304-1308. DOI:10.1126/science.aaf6635 |
| [32] |
C. Le, Y. Liang, R.W. Evans, X. Li, D.W.C. MacMillan, Nature 547 (2017) 79-83. DOI:10.1038/nature22813 |
| [33] |
J. Ye, I. Kalvet, F. Schoenebeck, T. Rovis, Nat. Chem. 10 (2018) 1037-1041. DOI:10.1038/s41557-018-0085-9 |
| [34] |
M.A. Ashley, C. Yamauchi, J.C.K. Chu, et al., Angew. Chem. Int. Ed. 58 (2019) 4002-4006. DOI:10.1002/anie.201812227 |
| [35] |
H. Tian, Q. Xia, Q. Wang, et al., Org. Lett. 21 (2019) 4585-4589. DOI:10.1021/acs.orglett.9b01491 |
| [36] |
J. Wang, B. Huang, Y. Gao, C. Yang, W. Xia, J. Org. Chem. 84 (2019) 6895-6903. DOI:10.1021/acs.joc.9b00708 |
| [37] |
X. Zhang, D.W.C. MacMillan, J. Am. Chem. Soc. 139 (2017) 11353-11356. DOI:10.1021/jacs.7b07078 |
| [38] |
S. Mkrtchyan, V.O. Iaroshenko, J. Org. Chem. 85 (2020) 7152-7174. DOI:10.1021/acs.joc.0c00537 |
| [39] |
M.D. Vu, M. Das, X.W. Liu, Chem. Eur. J. 23 (2017) 15899-15902. DOI:10.1002/chem.201704224 |
| [40] |
R. Zhou, Y.Y. Goh, H. Liu, et al., Angew. Chem. Int. Ed. 56 (2017) 16621-16625. DOI:10.1002/anie.201711250 |
| [41] |
J. Hou, A. Ee, H. Cao, et al., Angew. Chem. Int. Ed. 57 (2018) 17220-17224. DOI:10.1002/anie.201811266 |
| [42] |
W.L. Yu, Y.C. Luo, L. Yan, et al., Angew. Chem. Int. Ed. 58 (2019) 10941-10945. DOI:10.1002/anie.201904707 |
| [43] |
W.K. Weigel Ⅲ, H.T. Dang, H.B. Yang, D.B.C. Martin, Chem. Commun. (Camb.) 56 (2020) 9699-9702. DOI:10.1039/D0CC02804E |
| [44] |
H.B. Yang, A. Feceu, D.B.C. Martin, ACS Catal. 9 (2019) 5708-5715. DOI:10.1021/acscatal.9b01394 |
| [45] |
H. Wang, Y. Gao, C. Zhou, G. Li, J. Am. Chem. Soc. 142 (2020) 8122-8129. DOI:10.1021/jacs.0c03144 |
| [46] |
I. Bosque, T. Bach, ACS Catal. 9 (2019) 9103-9109. DOI:10.1021/acscatal.9b01039 |