 2021, Vol. 32
2021, Vol. 32 


b The Key Laboratory of Water and Sediment Sciences, Ministry of Education, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China;
c School of Chemistry and Beijing Advanced Innovation Center for Biomedical Engineering, Beihang University, Beijing 100191, China;
d State Environmental Protection Key Laboratory of All Material Fluxes in River Ecosystems, Peking University, Beijing 100871, China
With the extensive use of drugs and cosmetics, pharmaceutical and personal care products (PPCPs) have drawn more and more attention for their potential harm to human body and ecology. PPCPs have been detected in sewage treatment plants, rivers, lakes and groundwater in some regions, due to the development of environmental monitoring technology and scientists' in-depth study of environmental pollution [1, 2]. Acetaminophen (ACE) [3, 4], as a typical representative of PPCPs, is widely used in analgesic and antipyretic drug to relieve fever, headache and mild pain, whereas it is difficult to be removed rapidly in the environment [5]. Although the detected concentration ranges from ng/L to μg/L, they will enter the environment by various ways and accumulate through the biological amplification of the food chain, and finally endanger the aquatic environment and human health, embodying in abnormal physiological processes and reproductive disorders. Considering effective degradation of ACE is hard to be achieved by the traditional wastewater treatment processes, it is particularly important to develop advanced environmentally-friendly technology.
Advanced oxidation processes (AOPs), as one of the wastewater treatments, can degrade the contaminants by generating the reactive oxygen species (ROS) [6, 7]. Sulfate radicals (SO4*−) [8, 9] based AOPs comparison with hydroxyl radicals (*OH) [10] possess long half-life, high selectivity, and wide pH range [11], which are generated through using ultraviolet (UV) radiation, ultrasound, heat treatment, or catalysts activated persulfate (PS) or peroxymonosulfate (PMS) process [12-14]. This is reported that heterogeneous catalysts (Co, Fe, Cu, Fe-Co, MnFe2O4, CoFe2O4, etc.) [15-17] have been applied in catalytic oxidation of contaminants by activation of PS or PMS [18]. Especially, cobalt-based materials are evaluated as high-efficiency catalysts by activation of PMS [19]. However, the works mainly focus on crystalline Co(OH)2 and cobalt-based materials as PMS activator [20], the amorphous ones are rarely reported. Amorphous catalysts exhibited disordered atom arrangement, high structural flexibility, and abundant inherent defects, which have shown attractive prospects in environmental and energy-related application [21].
Herein, we have explored amorphous Co(OH)2 nanocages as PMS activator for efficient degradation of ACE. The initial pH is one of the critical influence factors on remove ACE. The radicals quenching experiments were designed to confirm the primary active radicals, which also identified by electron paramagnetic resonance (EPR) analysis. Besides, based on the form intermediates, we propose the ACE removal pathway via the amorphous Co(OH)2 by activating PMS. Finally, the mechanism of ACE degradation was elucidated by the analysis of degradation intermediates and theoretical calculation, indicating that the electrophilic SO4*− and HO* tend to attack the atoms of ACE with high Fukui index (f−). Our finding highlights the remarkable advantages of amorphous materials as heterogeneous catalysts in sulfate radicals-based AOPs and sheds new lights on water treatment for the degradation of emerging contaminants.
Amorphous Co(OH)2 nanocages were developed in a coordinating etching and precipitating process following our previous work with minor modification [22]. Firstly, we prepared octahedral Cu2O based on our previous work [23]. Typically, 0.171 g CuCl2·2H2O and 4.5 g polyvinylpyrrolidone (PVP) were dissolved in the aqueous solution, then 10 mL NaOH (2.0 mol/L) was added into the mixed solution. 10 mL ascorbic acid (0.6 mol/L) was dropwise added after 30 min in reaction solution. The turbid red Cu2O was formed after 3 h stirring. All the process was carried out under constant stirring and heated in a water bath at 55 ℃. The obtained samples was washed with H2O and ethanol, and dried at 60 ℃ in vacuum. As to the synthesis of amorphous Co(OH)2 nanocages, 17 mg of CoCl2·H2O and 50 mg Cu2O were added to the mixed solvent of 50 mL ethanol and 50 mL H2O with 3.33 g PVP. 40 mL Na2S2O3 (1.0 mol/L) solution was dropwise added into the uniform solution with stirring at room temperature after sonication. The color of suspension solution changed to pale green in 5 min. The amorphous Co(OH)2 nanocages were obtained after washing with H2O (2 times) and ethanol (3 times), and being dried at 60 ℃ in vacuum. The materials were characterized by transmission electron microscopy (TEM), selected area electron diffraction (SAED), high-resolution TEM (HRTEM), environmental scanning electron microscopy (ESEM), powder X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). Chemical regents, characterizations, and formation mechanism were shown in Texts S1 and S2 (Supporting information).
Amorphous Co(OH)2 nanocages as PMS activator for efficient degradation of ACE were performed. The details are as follows: adding ACE (10 μmol/L) and PMS (50 μmol/L) into 100 mL H2O with adjusting pH 5.0±0.1, the 0.1 g/L catalyst was added into the above solution. Collected 1 mL reaction solution in liquid phase vial filtered by NYLON 0.22 μm syringe filter and then quickly mixed with 100 μL Na2S2O3 (100 mmol/L) solution to quench the active radicals at different sampling time. The high-performance liquid chromatography (HPLC) was carried out on the Agilent 1260 Infinity Ⅱ to test the concentration of ACE. The products of formation were identified by ultra-high performance liquid chromatography (UHPLC) via electrospray ionization (ESI) source on Thermo Scientific. Testing information was described in Test S3 (Supporting information). The initial pH is one of the critical influences on remove ACE, designing the initial pH from 3 to 11. We also study the quenching experiments with addition of tert-butanol (TBA, 50 mmol/L) and ethanol (EtOH, 50 mmol/L) into the degradation system to quench*OH and all radicals, respectively [24]. EPR analysis was employed 5, 5-dimethyl-1-pyrrolineN-oxide (DMPO) as trapping agent for the generated radicals via PMS activation system by adding catalyst. The radicals attacking mechanism of ACE degradation was speculated by the analysis of generated intermediates via the density-functional theory (DFT) calculation in Text S4 (Supporting information) [25].
Figs. 1a and b exhibit the ESEM image of amorphous Co(OH)2 with octahedral cage-like nanostructures, possessing an edge length of 600−800 nm. These nanocages perfectly inherited the dimension and shape of the pre-grown Cu2O templates (Fig. S1a in Supporting information). The TEM images disclose the flake-like secondary structures on the surface of the amorphous Co(OH)2 nanocages, which are beneficial to the increase of specific surface area (Figs. 1c and d) [26, 27]. The corresponding SAED image (inset in Fig. 1d) with only halo suggests that it is amorphous state, which is also proved by HRTEM image (Fig. 1e). Commercial crystal β-Co(OH)2 was employed as a counterpart. The corresponding TEM and ESEM images in Fig. 1f and Fig. S1b (Supporting information) demonstrate that it possesses hexagonal sheet structure and crystal textures. The lattice spacing of 0.465 nm is assigned to (001) crystal planes of β-Co(OH)2 (inset in Fig. 1f).

|
Download:
|
| Fig. 1. Amorphous Co(OH)2 nanocages of (a) and (b) ESEM images; (c) and (d) TEM images, while the inset is the SAED pattern; (e) HRTEM image. (f) TEM images of commercial crystal β-Co(OH)2. | |
{kind=link}
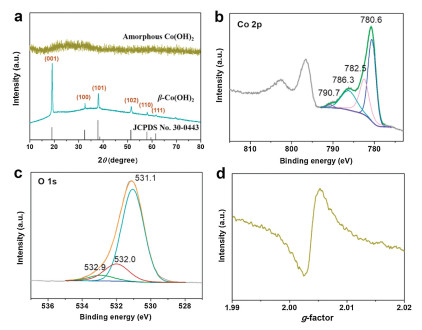
Further structural characterization and chemical state analysis were performed on amorphous Co(OH)2 nanocages. In Fig. 2a, none sharp Bragg reflection peak can be observed, which was consistent with the TEM results. The commercial Co(OH)2 was coincided with hexagonal cell of brucite-like β-Co(OH)2 with the crystalline diffraction peaks (JCPDS No. 30-0443) [28]. The chemical states of amorphous Co(OH)2 nanocages (Figs. 2b and c) and crystalline β-Co(OH)2 (Fig. S2, Supporting information) were measured by XPS analysis, setting C 1s at binding energy of 284.8 eV as reference. In Fig. 2b in two main peaks of Co2p3/2 and Co 2p1/2 were located at binding energy of 780.6 and 796.7eV, respectively, are together with two shake-up satellite peaks, which are characteristic features of Co2+ [29]. Co 2p3/2 spectrum was fitted with four peaks at 780.6, 782.5, 786.3 and 790.7eV, which agree with Co 2p3/2 signals from cobalt hydroxide species reported in literature [28]. Meanwhile, we also observed the high-resolution O 1s spectrum three peaks in Fig. 2c. The main O 1s peak at 531.1 eV is consistent with O that from cobalt hydroxides [30]. The peaks at 532.0 eV and 532.9 eV stand for the presence of oxygen vacancies and structural water [30]. For providing more evidence for the existence of oxygen vacancies, we carried out EPR analysis. As shown in Fig. 2d, a clear signal in EPR measurement at g=2.004 can demonstrate the existence of oxygen vacancies in amorphous Co(OH)2 [31]. These results indicate that the synthesized amorphous sample is cobalt hydroxide with oxygen vacancies.

|
Download:
|
| Fig. 2. Amorphous Co(OH)2: (a) XRD patterns; XPS spectra of (b) Co 2p and (c) O 1s spectrum; (d) EPR spectrum. | |
{kind=link}
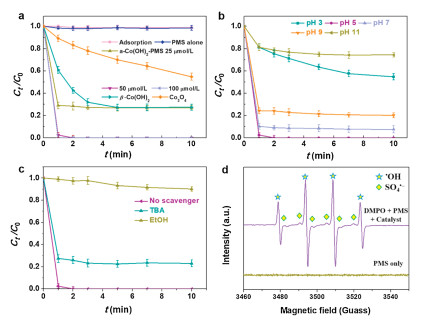
We have designed the amorphous Co(OH)2 as PMS activator for efficient degradation of ACE in Fig. 3a. In PMS alone reaction system not adding the catalyst, we can find PMS was hardly removal ACE, because the ACE content was almost same at different sampling time. In amorphous Co(OH)2 only, the adsorption experiment of ACE in 10 min was able to ignore for ACE degradation. When adding the amorphous Co(OH)2 into PMS system the activating removal of ACE was 97.5% within 1 min, corresponding the pseudo-first order kinetic rate constant (k1 = 3.68 min−1). The different PMS dosage and catalyst-PMS systems of rate constants for ACE removal were shown in Fig. S3 (Supporting information) [32, 33], realizing completely degradation of ACE by increasing the reaction time. In contrast, the crystalline Co(OH)2 for the ACE removal with the efficiency was 72.6% (k1 = 0.5 min−1). The efficiency of Co3O4 as benchmark catalyst on ACE removal was operated via activating PMS, which the degradation of ACE was 45.2% with k1 = 0.11 min−1. In addition, the influence of adding different PMS concentration for the ACE removal was investigated. When change the PMS dosage from 25 μmol/L to 100 μmol/L with the catalyst, the ACE removal rate was increased significantly, especially addition amount of 50 and 100 μmol/L realized completely removal of ACE within 1−2 min. The kinetic rate constant of 50 μmol/L was triple than that of 25 μmol/L. As considered from environment protection, we used 50 μmol/L PMS as the optimum dosage in the subsequent experiments. So the amorphous Co(OH)2 nanocages have higher catalytic effect than crystalline Co(OH)2 and Co3O4, because of the following reasons [27]: 1) Amorphous materials with hollow structures possess high specific surface areas with more reactive sites to participate in the catalytic redox reactions; 2) abundant defect/vacancies structures caused by amorphous catalysts that result in unusual enhancement in catalytic activities; 3) composition flexibility and disorder atomic arrangement with lots of vacant sites and active sites better for molecular diffusion and reaction progress [21]. Besides, they can provide numerous active sites make the Co2+ from amorphous Co(OH)2 to form CoOH+ as the key species to activated PMS [34, 35]. Considering the problems of leaching cobalt ions and environmental protection, the leaching mass ratio is 1.4% with completely removal of ACE via PMS activation. And the leaching concentration of Co2+ is 0.9 mg/L, which is below the national discharge standards (1.0 mg/L). It follows that the obtained amorphous Co(OH)2 nanocages were suitable for ACE removal via PMS activation.

|
Download:
|
| Fig. 3. (a) Different PMS dosage and catalyst-PMS systems for the ACE removal. (b) Effect of initial pH. (c) Effect of different scavenger. (d) EPR spectra in PMS only and amorphous Co(OH)2-PMS system with DMPO as trapping agent. | |
{kind=link}
The dissociation of pollutants molecule and formation of active radicals strongly depend on the pH effect [20, 36]. The initial pH is one of the critical influences on remove ACE, designing the initial pH from 3 to 11 as depicted in Fig. 3b and Fig. S4a (Supporting information). The ACE removal rate was from 45.3%-100%, and then to 92.4% with adjusting pH from 3 to 7 within 10 min. The corresponding reaction rate constantk under pH of 5 was 17.5 times to that pH of 3 (k1 = 0.21 min−1). Then adjusting pH from 9 to 11, the degradation efficiency dropped off from 79.9%-25.7%. The pH variation under optimal pH 5 reaction system was monitored. When the reaction times in 2 min, the pH changed from 5.0-5.2±0.1. When prolonged the reaction time to 10 min, the pH changed to 6.2±0.1, and remained stationary about 5min the pH almost unchanged. Considering the alkaline activation contribute to the ACE removal, the experiment of pure PMS and ACE under pH 10 without catalyst was also performed as shown in Fig. S4b (Supporting information), whose removal efficiency was reduced about 5.1% via PMS activation in strong alkali system. Besides, the formation of radical types of PMS (pK = 9.4) rely on initial pH effect. At the neutral and acid conditions, a large number of HSO5− were generated by PMS activation. The key species of CoOH+ and HSO5− react with each other to form SO4*− radicals. Subsequently, the *OH was obtained through the reaction of HSO5−/SO4*− and OH−/H2O [16]. The generation of SO4*− and *OH attacking ACE molecule result in high-efficiency removal. Under strong acid about pH < 3, the key species of CoOH+ was suppressed, leading to the degradation of ACE fall down [6]. But under the condition of pH > 9, numerous of SO52- ions were formed via activation process, whose oxidizing ability was lower than HSO5− [36]. Under strong alkali condition, HO* as the prominent radicals were weaker than SO4*−, leading to poorly redox ability for ACE removal [37]. The ACE existing types as a function of pH were drawn and discussed in Fig. S5 (Supporting information). Thus, the optimum pH was 5, which was also consistent with the previous studies that the optimal range of pH for cobalt-PMS system was 4-9 [38].
Radicals quenching experiments were then conducted to evaluate the dominant radicals during ACE removal using amorphous Co(OH)2 as PMS activator. Basically, TBA and EtOH were added during the reaction to quench the *OH and both radicals (*OH and SO4*−), respectively [6, 24] (Fig. 3c and Fig. S6 in Supporting information). The k1 values decreased from 3.68 min−1 in the absence of scavengers to 1.30 and 0.02 min−1 within 1min in the presence of 50 mmol/L TBA or EtOH, respectively, indicating both radicals contributed to the ACE degradation while SO4*− radicals played more important role. In addition, EPR was employed to confirm the generation of SO4*− and *OH with the addition of DMPO as trapping agent. No characteristic peak was observed in the PMS only system in Fig. 3d, indicating that no active species signals were captured without the spin-trapping agent. When adding DMPO into PMS solution, strong EPR signals can be assigned to the characteristic signals of 5, 5-dimethylpyrroline-(2)-oxyl-(1) (DMPOX, the nitroxide radical of DMPO) [20]. After adding amorphous Co(OH)2 as PMS activator with addition of DMPO, the characteristic of DMPO-*OH signals [39] and a six-line signal of DMPO-SO4*− adducts were detected [40, 41]. The EPR results confirmed the generation of SO4*− and *OH radicals in the amorphous Co(OH)2-PMS activation system, which is in line with the results of quenching experiments.
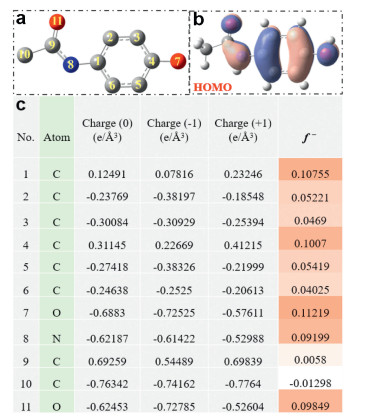
The radicals attacking mechanism of ACE degradation was speculated by the analysis of generated intermediates via DFT calculation as drawn in Figs. 4a-c. The electrophilic SO4*− and *OH [8] inclined to attack active sites of the highest occupied molecular orbital (HOMO) of ACE (Fig. 4b), which easily lose electron and readily escape of electron [25]. Meanwhile, the Fukui index (f−) and natural population analysis (NPA) charge distribution of ACE were listed in Fig. 4c. The higher f − value of site on ACE molecule were more easily to lose electron and be attacked by radicals. Four proposed ACE pathways via activating PMS by amorphous Co(OH)2 as catalysts were depicted in Scheme 1. The chemical formulas of intermediates and transformation products (TPs) of ACE identified in PMS activation system by UHPLC were displayed in Table S1 (Supporting information). (1) For pathway Ⅰ, two isomers of ACE through the coupling are identified as the dimer intermediates (m/z 301.1179) [3], as previously reported by Chen et al. ACE dimer is assumed to be formed through the radical coupling of ACE phenoxyl radical (ACE*), which is generated through radicals induced one electron oxidation of ACE [4]. Then loss of one of the acetamide groups followed by hydroxylation, resulting in the generation of m/z 260.0914 [3]. (2) In pathway Ⅱ and Ⅲ, the SO4*− and *OH attack the N-C bond of ACE make bond cleavage to generate the following products, e.g., 4-aminophenol (m/z 109.97744), acetic acid (m/z 61.04042), acetamide (m/z 60.04520), hydroquinone (m/z 111.11703) [42], which was matched with higher Fukui index of N8 (f − = 0.09199) and C1 (f − = 0.10755). The products m/z 109.10143 and m/z 119.01904 were produced through further hydroxylation. (3) For pathway Ⅳ, the aniline ring of ACE via direct oxidation by hydroxylation to obtain the products m/z 167.98244 after *OH addition on the sites of C5 (f − = 0.05419). Then suffering the ring opening or N-C bond cleavage result in the generation of m/z 200.20053 and 126.05492 [43]. Finally, the low molecular intermediates (e.g., carboxylic acids, acetic acids, formic acids) future oxidized by active radicals and mineralized to generate the products of CO2 and H2O [44]. The toxicity of the intermediates, residual PMS, and radicals were discussed and depicted in Fig. S7 (Supporting information).

|
Download:
|
| Fig. 4. Acetaminophen (ACE): (a) Chemical structure of ACE molecular; (b) HOMO; (c) the Fukui index () and NPA charges. | |
{kind=link}

|
Download:
|
| Scheme 1. Proposed the pathway of ACE via activating PMS by amorphous Co(OH)2 as catalyst. | |
{kind=link}
In conclusion, high active amorphous Co(OH)2 nanocages were investigated as PMS activator for efficient removal of ACE. In this system, ACE can be completely degraded within 2 min at optimum pH 5 with reaction rate constant of 3.68 min−1, which was much better than that of crystalline β-Co(OH)2 and Co3O4. The superior performance of our amorphous materials for PMS activation can be attributed to the hollow architecture, large specific surface area, more reactive sites, and abundant oxygen vacancies structures, benefitting the catalytic redox reactions between catalyst and PMS. The radicals quenching experiment demonstrated that SO4*− radicals induced by amorphous Co(OH)2 play an important role in ACE degradation rather than that *OH radicals. Intermediates analysis and DFT calculation indicate that the atoms of ACE with high Fukui index (f−) were more prone to be attacked by SO4*− and *OH. Our work demonstrates the promising prospects of amorphous materials as catalysts in water treatment and deepen the understanding of reactive mechanism for degradation of emerging organic contaminants.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial supports from China Postdoctoral Science Foundation (Nos. 2019M650007 and 2020M670088) and National Natural Science Foundation of China (Nos. 21906001, 51721006) are highly acknowledged. We also thank the Beijing Nova Program (No. Z19111000110000) supported this work.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.11.026.
| [1] |
Z. Cai, A.D. Dwivedi, W.N. Lee, et al., Environ. Sci.:Nano 5 (2018) 27-47. DOI:10.1039/C7EN00644F |
| [2] |
H. Ji, P. Du, D. Zhao, et al., Appl. Catal. B 263 (2020) 118357. DOI:10.1016/j.apcatb.2019.118357 |
| [3] |
M. Penas-Garzon, A. Gomez-Aviles, C. Belver, J.J. Rodriguez, J. Bedia, Chem. Eng. J. 392 (2020) 124867. DOI:10.1016/j.cej.2020.124867 |
| [4] |
Y. Chen, X. Zhang, L. Mao, Z. Yang, Chem. Eng. J. 330 (2017) 1091-1099. DOI:10.1016/j.cej.2017.07.148 |
| [5] |
L. Chen, H.D. Ji, J.J. Qi, et al., Chem. Eng. J. 406 (2021) 126877. DOI:10.1016/j.cej.2020.126877 |
| [6] |
R. Zhang, Y. Wan, J. Peng, et al., Chem. Eng. J. 372 (2019) 796-808. DOI:10.1016/j.cej.2019.04.188 |
| [7] |
Y. Du, W. Ma, P. Liu, B. Zou, J. Ma, J. Hazard. Mater. 308 (2016) 58-66. DOI:10.1016/j.jhazmat.2016.01.035 |
| [8] |
M. Ma, L. Chen, J. Zhao, W. Liu, H. Ji, Chin. Chem. Lett. 30 (2019) 2191-2195. DOI:10.1016/j.cclet.2019.09.031 |
| [9] |
M. Zhang, J. He, Y. Chen, et al., Chin. Chem. Lett. 31 (2020) 2721-2724. DOI:10.1016/j.cclet.2020.05.001 |
| [10] |
A. Zhu, Y. Guo, G. Liu, et al., Chin. Chem. Lett. 30 (2019) 2241-2244. DOI:10.1016/j.cclet.2019.09.003 |
| [11] |
C. Liu, L. Liu, X. Tian, et al., Appl. Catal. B 255 (2019) 117763. DOI:10.1016/j.apcatb.2019.117763 |
| [12] |
Q. Yang, H. Choi, Y. Chen, D.D. Dionysiou, Appl. Catal. B 77 (2008) 300-307. DOI:10.1016/j.apcatb.2007.07.020 |
| [13] |
M. Nie, Y. Yang, Z. Zhang, et al., Chem. Eng. J. 246 (2014) 373-382. DOI:10.1016/j.cej.2014.02.047 |
| [14] |
C. Tan, D. Fu, N. Gao, et al., J. Photochem. Photobiol. A 332 (2017) 406-412. DOI:10.1016/j.jphotochem.2016.09.021 |
| [15] |
J. Deng, S. Feng, X. Ma, et al., Sep. Purif. Technol. 167 (2016) 181-189. DOI:10.1016/j.seppur.2016.04.035 |
| [16] |
P. Hu, M. Long, Appl. Catal. B 181 (2016) 103-117. DOI:10.1016/j.apcatb.2015.07.024 |
| [17] |
C. Cai, H. Zhang, X. Zhong, L. Hou, J. Hazard. Mater. 283 (2015) 70-79. DOI:10.1016/j.jhazmat.2014.08.053 |
| [18] |
J. Wang, S. Wang, Chem. Eng. J. 334 (2018) 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
| [19] |
P. Shao, J. Tian, X. Duan, et al., Chem. Eng. J. 359 (2019) 79-87. DOI:10.1016/j.cej.2018.11.121 |
| [20] |
X. Tao, P. Pan, T. Huang, et al., Chem. Eng. J. 395 (2020) 125186. DOI:10.1016/j.cej.2020.125186 |
| [21] |
S. Anantharaj, S. Noda, Small 16 (2020) 1905779. DOI:10.1002/smll.201905779 |
| [22] |
J. Nai, Y. Tian, X. Guan, L. Guo, J. Am. Chem. Soc. 135 (2013) 16082-16091. DOI:10.1021/ja402751r |
| [23] |
J.J. Podesta, R.C.V. Piatti, A.J. Arvia, et al., Int. J. Hydrogen Energy 17 (1992) 9-22. DOI:10.1016/0360-3199(92)90216-J |
| [24] |
X. Lei, M. You, F. Pan, et al., Chin. Chem. Lett. 30 (2019) 2216-2220. DOI:10.1016/j.cclet.2019.05.039 |
| [25] |
X. Zhao, P. Du, Z. Cai, et al., Environ. Pollut. 232 (2018) 580-590. DOI:10.1016/j.envpol.2017.09.094 |
| [26] |
X. Guan, J. Nai, Y. Zhang, et al., Chem. Mater. 26 (2014) 5958-5964. DOI:10.1021/cm502690u |
| [27] |
J. Liu, J. Nai, T. You, et al., Small 14 (2018) 1703514. DOI:10.1002/smll.201703514 |
| [28] |
J. Yang, H. Liu, W.N. Martens, R.L. Frost, J. Phy. Chem. C 114 (2010) 111-119. DOI:10.1021/jp908548f |
| [29] |
A. Meng, L. Sheng, K. Zhao, Z. Li, J. Mater. Chem. B 5 (2017) 8934-8943. DOI:10.1039/C7TB02482G |
| [30] |
J.K. Chang, C.M. Wu, I.W. Sun, J. Mater. Chem. 20 (2010) 3729-3735. DOI:10.1039/b925176f |
| [31] |
Y. Chen, Y. Guo, H. Hu, et al., Inorg. Chem. Commun. 82 (2017) 20-23. DOI:10.1016/j.inoche.2017.05.005 |
| [32] |
Q. Chen, L. Chen, J. Qi, et al., Chin. Chem. Lett. 30 (2019) 1214-1218. DOI:10.1016/j.cclet.2019.03.002 |
| [33] |
H. Ji, Y. Zhu, J. Duan, W. Liu, D. Zhao, Chin. Chem. Lett. 30 (2019) 2163-2168. DOI:10.1016/j.cclet.2019.06.004 |
| [34] |
P. Shi, X. Dai, H. Zheng, et al., Chem. Eng. J. 240 (2014) 264-270. DOI:10.1016/j.cej.2013.11.089 |
| [35] |
Q. Yang, H. Choi, D.D. Dionysiou, Appl. Catal. B 74 (2007) 170-178. DOI:10.1016/j.apcatb.2007.02.001 |
| [36] |
M. Xu, J. Li, Y. Yan, et al., Chem. Eng. J. 369 (2019) 403-413. DOI:10.1016/j.cej.2019.03.075 |
| [37] |
C. Tan, N. Gao, Y. Deng, et al., J. Hazard. Mater. 276 (2014) 452-460. DOI:10.1016/j.jhazmat.2014.05.068 |
| [38] |
G.P. Anipsitakis, D.D. Dionysiou, M.A. Gonzalez, Environ. Sci. Technol. 40 (2006) 1000-1007. DOI:10.1021/es050634b |
| [39] |
Y. Wang, S. Indrawirawan, X. Duan, et al., Chem. Eng. J. 266 (2015) 12-20. DOI:10.1016/j.cej.2014.12.066 |
| [40] |
G.D. Fang, D.D. Dionysiou, S.R. Al-Abed, D.M. Zhou, Appl. Catal. B 129 (2013) 325-332. DOI:10.1016/j.apcatb.2012.09.042 |
| [41] |
Y. Chen, G. Zhang, H. Liu, J. Qu, Angew. Chem. Int. Ed. 58 (2019) 8134-8138. DOI:10.1002/anie.201903531 |
| [42] |
M. Noorisepehr, B. Kakavandi, A.A. Isari, et al., Sep. Purif. Technol. 250 (2020) 116950. DOI:10.1016/j.seppur.2020.116950 |
| [43] |
B. Li, X. Ma, Q. Li, et al., Chem. Eng. J. 392 (2020) 124552. DOI:10.1016/j.cej.2020.124552 |
| [44] |
H. Zhang, Q. Ji, L. Lai, G. Yao, B. Lai, Chin. Chem. Lett. 30 (2019) 1129-1132. DOI:10.1016/j.cclet.2019.01.025 |