 2021, Vol. 32
2021, Vol. 32 

In the past decades, DNA nanotechnology has developed rapidly and many DNA nanostructures are fabricated for drug delivery [1], live cell imaging [2], diagnosis [3], and so on. Excellent biocompatibility, inherent biodegradability, as well as versatile programmability, enable DNA nanostructures to be predominant platforms for cancer therapy [1, 4, 5]. The changes in the tumor microenvironment, including pH and redox, are utilized to construct stimuli-responsive drug nanocarriers [6, 7]. In particular, pH-responsive materials are valuable for the fabrication of DNA-based drug nanodevices. The i-motif structure, consisting of a cytosine-rich single-stranded DNA sequence, can be folded into four-stranded (C-quadruplex) under the adjustment of environmental pH from 7.4 to 5.0 [8]. Based on this characteristic, the i-motif structures are widely employed as pH-responsive drug delivery systems (DDSs) [8, 9].
To enhance cancer therapeutic efficiency, photothermal therapy (PTT) is exploited to combine chemotherapy [10, 11]. Nanomaterials-based PTT converts the near-infrared (NIR) irradiation into heat for the ablation of cancer cells, which has shown the potential in cancer therapy [10-12]. Owing to the excellent photothermal conversion efficiency, gold nanoparticles (AuNPs) become representative photothermal agents for PPT [13]. DNA structures (such as targeting ligand (aptamer)) are explored to integrate into AuNPs for the fabrication of multifunctional DDSs which can enhance the effect of PTT [14, 15]. Generally, the DNA structure can conjugate with AuNPs via the Au-S bond [16]. It is worth noting that the charge repulsion of negatively charged AuNPs and negatively charged DNA makes this process difficult. Then, a salt-aging method was developed to shield charge repulsion, which took two full days to slowly add salt in solution to avoid aggregation of AuNPs [17, 18].
Great efforts were paid to simplify the complex procedure, for instance, a freeze-thaw method was explored to efficiently accelerate the process [19]. The freeze-thaw method could be summarized as fast freezing the mixtures of DNA and AuNP and then slowly thawing it at room temperature. In addition, the freeze-thaw method increases the salt concentration around the AuNPs by freezing rather than adding more salt, and a high local salt concentration is attributed to the crystallization of water molecules [19]. It is well-known that colloid nanoparticles produce irreversible aggregation after being frozen, and this aggregation resulting from the enhanced salt concentration screen charge repulsion between nanoparticles [20]. The stability and dispersity of frozen AuNPs were remarkably enhanced after the modification of DNA sequences. Herein, the thiolated DNA sequences can be easily assembled on AuNPs via a freeze-thaw cycle.
Doxorubicin (Dox), a common anticancer drug, can be noncovalently intercalated into GC base pairs of DNA nanostructures [21], and the drug loading capability can be tailorable by adjusting the GC content of DNA nanostructures [22]. Thus, the high content of GC base pairs in the complementary structure of DNA strands with i-motif guarantees high drug-loading capability. MUC1 protein is a transmembrane glycoprotein that overexpressed on many epithelial cancers (e.g., breast, lung, and pancreatic cancer) and serves as an entry portal for its aptamer [23, 24]. To enhance specifically cellular uptake, MUC1 aptamer (that bind ligands to the MUC1 glycoprotein tumor marker) is chosen as targeting ligand for cancer therapy [24, 25].
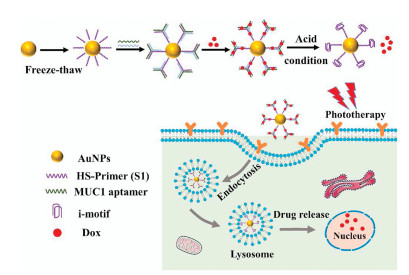
In this article, we developed a multifunctional drug nanocarrier for synergistic photothermal-chemotherapy. The addition of the i-motif structure realized the characteristics of pH-responsive and provided the high content of GC base pairs for Dox-loading. As shown in Scheme 1, the thiolated single-stranded DNA (S1), containing the i-motif sequence, was successfully assembled on AuNPs via a freeze-thaw method to obtain the AuNP-S1. To enhance the tumor-targeting capability, MUC1 aptamers were selected as targeting ligands to recognize the MUC1 protein overexpressing on the human lung cancer cell line (A549). MUC1 aptamer was covalently connected with strands S2 and S3, then the strands S2 and S3 would hybridize with strand S1 on AuNP-S1 to form a complex structure (Y-shape DNA). The non-targeting strand (NEG) was selected as the negative control of MUC1 aptamer, which was connected with strand S4 and S5. At the same time, the strands S1, S4, and S5 were assembled into the Y-shape-NEG structure. The DNA sequences required in this article are shown in Table S1 (Supporting information).

|
Download:
|
| Scheme 1. Schematic illustration of targeted pH-responsive i-motif DNA-conjugated gold nanoparticles for synergistic photothermal-chemotherapy. | |
{kind=link}
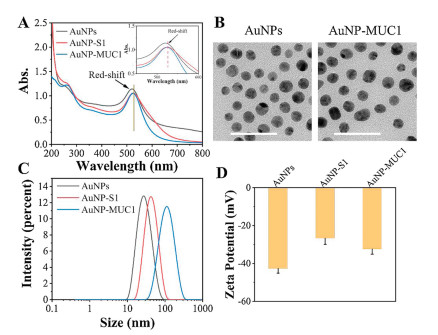
To investigate whether the DNA strands were successfully assembled on AuNPs. The UV–vis absorption spectrum showed that the characteristic absorption peak of AuNPs was 520 nm, while the AuNP-S1 and AuNP-MUC1 showed the absorption band at 527 nm and 529 nm, respectively (Fig. 1A). The red-shift confirmed the successful modification of AuNPs by DNA structure. Transmission electron microscopy (TEM) images revealed that the AuNPs were kept in a dispersive with an average size of 17.57±1.54 nm (Fig. 1B). Dynamic light scattering (DLS) showed that the average hydrodynamic diameter of AuNPs was about 25 nm. The size was increased to 42 nm with strand S1 modification and further increased to 107 nm after the hybridization with strands S2 and S3 (Fig. 1C). The zeta potential of naked AuNPs was about −43 mV, while the zeta potential of AuNP-S1 changed to about −25 mV after modification with S1 (Fig. 1D). However, the negatively charged DNA modified on AuNPs did not decrease the zeta potential of AuNPs, which was caused by charge repulsion of Na+ and Mg2+ in solution [26]. After the grafting of strands S2 and S3, the zeta potential of AuNP-MUC1 was decreased to −33 mV. These results above indicated the functional DNA structures were successfully assembled on AuNPs.

|
Download:
|
| Fig. 1. (A) UV–vis absorption spectra, (B) TEM images, (C) hydrodynamic diameters, and (D) zeta potentials of AuNPs, AuNP-S1, and AuNP-MUC1. The scale bar represents 50 nm. | |
{kind=link}
The native polyacrylamide gel electrophoresis (Native PAGE) and agarose gel electrophoresis were used to confirm the assembly of AuNP-MUC1. Native PAGE (8%) demonstrated that the stands S1, S2, and S3 could be assembled into "Y-shape" DNA nanostructure via a "step-by-step" method, while the strands S1, S4, and S5 were assembled into "Y-shape-NEG" structure served as a negative control. As shown in Fig. 2A, the migration rate decreased gradually with the addition of DNA strands and the single clear band represented the high yield of the product. Also, agarose gel electrophoresis indicated that bare AuNPs easily self-aggregated in TAE buffer and were retained in the origin point due to the poor dispersity (Fig. 2B). AuNP-S1 showed a good dispersity and migrated from the origin point, suggesting the successful assembly of AuNP-S1. The color of AuNPs was turned from red to light blue after a freeze-thaw cycle, indicating irreversible aggregation of AuNPs, while the AuNP-S1 remained to be red (Fig. S1 in Supporting information). High stability of AuNP-S1 further verified that strands S1 were attached to AuNPs by a freeze-thaw process. Compared with AuNP-S1, AuNP-S1/S2 showed a lower migration rate which meant the assembly of strand S2 with AuNP-S1, and the migration rate of the band was ulteriorly decreased after incubation with stand S3 (Fig. 2B). In addition, the number of strand S1 modified on the AuNPs surface was evaluated by a standard linear calibration curve of FAM-labeled strand S1 (FAM-S1) [27]. AuNP-S1 was treated with DL-dithiothreitol (DTT) over 24 h to release FAM-S1, whose fluorescence maximums (measured at 520 nm, excited at 490 nm) were converted to molar concentration by a standard linear curve, and approximately 70 S1 strands were conjugated to one AuNP (Fig. S2 in Supporting information). Collectively, the above characterization experiments confirmed that Y-shape and Y-shape-NEG nanostructures were successfully assembled on AuNPs.

|
Download:
|
| Fig. 2. (A) Native PAGE analysis of different samples. M: DNA marker; line 1: S1; line 2: S1+S2; line 3: Y-shape; line 4: Y-shape-NEG. (B) Agarose gel electrophoresis of different samples. Line 5: bare AuNPs; line 6: AuNP-S1; line 7: AuNP-S1/S2; line 8: AuNP-MUC1; line 9: AuNP-NEG. (C) Rapid loading of Dox on AuNP-MUC1. (D) Fluorescence intensity of Dox with the addition of increased amounts of AuNP-MUC1 at pH 7.4 PBS. Molar ratios of AuNP-MUC1 to Dox were 0, 1×10−4, 2×10−4, 4×10−4, 8×10−4, 1×10−3, 2×10−3, 4×10−3, 8×10−3. | |
{kind=link}
To study the drug-loading efficiency of AuNP-MUC1, we used Dox as a model drug and utilized fluorescence spectrophotometer to quantify the loaded drugs. As shown in Fig. 2C, the fluorescence intensity of Dox was gradually reduced with the change of time, which was ascribed to the interaction between Dox and AuNP-MUC1. After 30 min, the fluorescence intensity no longer decreased, which meant that the drug-loading reached equilibrium. The fluorescence intensity of Dox would quench after the interaction with GC base pairs in AuNP-MUC1. With various concentrations of AuNP-MUC1 added to the free Dox solution, the fluorescence intensity decreased sharply. At a molar ratio (AuNP-MUC1:Dox) of 4×10−3, the fluorescence quenching reached saturation (Fig. 2D).
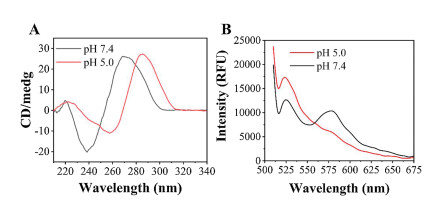
With regard to the pH-responsive characteristic of AuNP-MUC1, circular dichroism spectrum (CD) and fluorescence resonance energy transfer (FRET) were applied to verify the construction of the i-motif structure. As shown in Fig. 3A, the CD spectrum of Y-shape showed a positive peak at 268 nm and a negative peak at 237 nm at a pH of 7.4. When the pH decreased to 5.0, the positive peak and negative peak changed to 286 nm and 257 nm, respectively, which implied the formation of the i-motif structure [28]. FRET was not only utilized to verify that the Y-shape structure was successfully assembled in AuNPs, but also proved the pH-responsive characteristic of AuNP-MUC1. Respectively, we labeled strand S2 with the fluorescence donor FAM and strand S3 with fluorescence acceptor TAMRA. FRET efficiency is used to confirm the structural integrity of FAM/TAM/AuNP-MUC1 and inversely proportional to the donor-acceptor pair distance [29]. When the pH of the solution changed from 7.4 to 5.0, the FRET between donor and acceptor pair shifted from "on" to "off" state (Fig. 3B). FRET was the "on" state under the pH at 7.4, the fluorescence donor's emission led to the excitation of fluorescence acceptor and the fluorescence of the donor would be quenched at the same time, which results in high FRET efficiency (about 68%). The high FRET efficiency means the structural integrity of FAM/TAM/AuNP-MUC1. However, FRET was the "off" state under the pH at 5.0, the distance of the donor-acceptor pair significantly increased due to the pH-responsive of the i-motif structure, leading to low FRET efficiency (about 32%) and destroying the structural integrity of FAM/TAM/AuNP-MUC1. These results firmly confirmed that the multifunctional drug carrier was sensitive to the pH changes in the environment.

|
Download:
|
| Fig. 3. (A) Circular dichroism spectrum of Y-shape DNA under different pH values. (B) FRET assay of FAM and TAMRA double-labeled AuNP-MUC1 at different pH values (AuNP-MUC1 concentration: 4 nmol/L). | |
{kind=link}
Flow cytometry and confocal laser scanning microscope (CLSM) were exploited to examine the cellular uptake and drug-release of Dox@AuNP-NEG and Dox@AuNP-MUC1. As demonstrated in flow cytometry assay (Fig. 4A), the fluorescence intensity of Dox@AuNP-MUC1 was observed higher than that of Dox@AuNP-NEG due to the specific targeting of MUC1 aptamer. A549 cells were incubated with Dox@MUC1 for different time and analyzed by flow cytometry (Fig. 4B). Particularly, the mean fluorescence of the Dox@AuNP-MUC1 group gradually increased from 1 h to 4 h. But, the mean fluorescence was no longer enhanced when the incubation time reached 6 h, indicating the cellular uptake reached saturation. After incubation with Dox@AuNP-MUC1 and Dox@AuNP-NEG for different time (1, 2, and 4 h), A549 cells were washed twice with PBS and then stained with 4′, 6-diamidino-2-phenylindole (DAPI) to label nuclei, treated with Phalloidin FITC for actin cytoskeleton. Thereafter, the cellular uptake was further verified by CLSM in which the time-dependent fluorescence was increased from 1 h to 4 h (Fig. 4C). And the red fluorescence of Dox@AuNP-MUC1 treated group was observed stronger than that of Dox@AuNP-NEG treated group, which was consistent with the flow cytometry assay. These results demonstrated that the multifunctional nanocarrier could specifically target cancer cells.

|
Download:
|
| Fig. 4. (A) Flow cytometry assay of A549 cells incubated with free Dox, Dox@AuNP-NEG, and Dox@AuNP-MUC1. (B) Flow cytometry assay of A549 cells incubated with Dox@AuNP-MUC1 for different time (1, 2, 4, and 6 h). (C) CLSM images of A549 cells incubated with Dox@AuNP-NEG and Dox@AuNP-MUC1 for 1, 2, and 4 h, respectively, and stained with DAPI (blue fluorescence), Dox (red fluorescence), and actin (green fluorescence). Dox concentration: 2 μmol/L. The scale bar represents 20 μm and the concentration of AuNPs: 4 nmol/L. | |
{kind=link}
Moreover, to examine their stability, we carefully collected the cell culture medium (containing Dox@AuNP-NEG and Dox@AuNP-MUC1) after incubation at different time, then the samples were characterized by 1% agarose gel electrophoresis (Fig. S3 in Supporting information). Both two groups (Dox@AuNP-NEG and Dox@AuNP-MUC1) could keep stable within 4 h. To further demonstrate how long the nanocarriers can be stabilized in physiological conditions, the AuNP-MUC1 was incubated with the cell culture medium containing 10% fetal bovine serum (FBS) for different time (Fig. S4 in Supporting information). The degradation of nucleic acid in AuNP-MUC1 will increase its migration rate in agarose gel electrophoresis, and partial degradation was observed until 24 h, indicating their good stability in physiological conditions.
MTT assay was utilized to investigate the tumor inhibition efficiency of the nanocarrier. A549 cells were treated with free Dox, Dox@AuNP-NEG, and Dox@AuNP-MUC1 for 48 h with or without NIR irradiation (2 W/cm2, 4 min). The laser function power-dependent photothermal effect of Dox@AuNP-MUC1 was demonstrated by the photothermal heating curve, in which the temperature increment up to 14 ℃ at a power of 2 W/cm2 for 4 min (Fig. S5 in Supporting information). As shown in Fig. 5A, at Dox concentration of 6 μmol/L, the cell viability of Dox@AuNP-MUC1 (29% ± 2.4%) was lower than that of Dox@AuNP-NEG (35% ± 1.6%) due to the enhanced cellular uptake by MUC1 aptamers. Moreover, for the group treated with Dox@AuNP-MUC1 and 808 nm NIR irradiation (2 W/cm2, 4 min), the cell viability was dropped dramatically to 16% ± 2.0% at Dox concentration of 6 μmol/L due to the enhanced therapeutic efficiency induced by the photothermal effect of Dox@AuNP-MUC1. In this regard, this multifunctional drug nanocarrier exhibited obvious cytotoxicity toward A549 cells. To evaluate the cytotoxicity of drug carriers toward normal cells, Human umbilical vein endothelial cells (HUVECs) were incubated with Dox@AuNP-MUC1 with or without NIR irradiation for 48 h. As demonstrated in Fig. 5B, at Dox concentration 1.5 μmol/L, about 90% of cells were alive of both Dox@AuNP-MUC1 and NIR/Dox@AuNP-MUC1 group, indicating the noncytotoxicity of drug carrier toward normal cells. Besides, the NIR irradiation did not show an obvious influence on cell viability due to the lower MUC1 aptamer related cellular uptake on HUVECs cells. These results confirmed that Dox@AuNP-MUC1 nanocarrier combined with PTT exhibited significant anticancer efficiency and could distinguish cancer cells from healthy cells.

|
Download:
|
| Fig. 5. (A) Cell viability and (C) cell apoptosis of A549 cells after treatments with free Dox, Dox@AuNP-NEG, and Dox@AuNP-MUC1 for 48 h with or without NIR irradiation (2 W/cm2, 4 min). Q4-2 and Q4-4 were late apoptotic cells and early apototic cells. Data are presented as mean±SD (n=3). *P < 0.05, **P < 0.01, ***P < 0.001. (B) Cell viability of HUVECs incubated with Dox@AuNP-MUC1 for 48 h with or without NIR irradiation. The concentration of AuNPs: 4 nmol/L. | |
{kind=link}
As is known, both Dox (a therapeutic drug for cancer therapy) and moderate hyperthermia can induce cell apoptosis [30, 31]. Flow cytometry-based apoptosis assay was applied to verify the proliferation inhibition of cancer cells induced by apoptosis. A549 cells were treated with free Dox, Dox@AuNP-NEG, Dox@AuNP-NEG, and NIR/Dox@AuNP-MUC1 for 48 h. All groups contained an equivalent concentration of Dox (2 μmol/L). For the NIR/Dox@AuNP-MUC1 treated group, the cells were irradiated by NIR (808 nm, 2 W/cm2) for 4 min. As shown in Fig. 5C, the cells incubated with free Dox and Dox@AuNP-NEG exhibited apoptotic rates of 30% ± 2.0% and 43% ± 2.7% (sum of Q4−2 and Q4−4, Q4−4 represents early apoptosis and Q4−2 represents late apoptosis), respectively. The apoptotic rates were increased to 57% ± 3.2% and 80% ± 2.9% after treatment with Dox@AuNP-MUC1 and NIR/Dox@AuNP-MUC1, owing to MUC1 aptamer-mediated cellular internalization and increased temperature induced by NIR irradiation. These results suggested that the synergistic photothermal-chemotherapy exhibited much better therapeutic efficiency than chemotherapy alone.
In summary, we successfully synthesized a Dox-loaded i-motif DNA-conjugated AuNPs nanocarrier (Dox@AuNP-MUC1) for synergistic photothermal-chemotherapy. These novel platforms showed some attractive advantages: (1) The multifunctional DNA-AuNPs nanosystem was assembled from a facile freeze-thaw method, which exhibited good stability and high photothermal conversion efficiency; (2) The nanocarrier was stimuli-responsive to environmental pH due to the intercalation of pH-responsive i-motif structure; (3) Cancer cellular uptake was significantly and specifically enhanced by the modification of cell-targeting MUC1 aptamer; (4) Highly synergistic photothermal-chemotherapy was easily achieved by targeting pH-responsive nanoplatform-induced hyperthermia. In this study, the multifunctional nanocarrier was enabled to efficiently deliver anticancer drugs and selectively target cancer cells, thus showing the improved chemo-photothermal therapeutic efficiency. We believe that the multifunctional nanocomposites show the potential in synergistic photothermal-chemotherapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Sciences Foundation of China (No. 31971308) and National S&T Major Project (No. 2019ZX09301-147).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.058.
| [1] |
S. Li, Q. Jiang, S. Liu, et al., Nat. Biotechnol. 36 (2018) 258-264. DOI:10.1038/nbt.4071 |
| [2] |
Z. Di, J. Zhao, H. Chu, et al., Adv. Mater. 31 (2019) e1901885. DOI:10.1002/adma.201901885 |
| [3] |
Y. Peng, Y. Pan, Y. Han, et al., Anal. Chem. 92 (2020) 13478-13484. DOI:10.1021/acs.analchem.0c02906 |
| [4] |
Q. Mou, Y. Ma, F. Ding, et al., J. Am. Chem. Soc. 141 (2019) 6955-6966. DOI:10.1021/jacs.8b13875 |
| [5] |
J. He, T. Peng, Y. Peng, et al., J. Am. Chem. Soc. 142 (2020) 2699-2703. DOI:10.1021/jacs.9b10510 |
| [6] |
Z. Shi, Q. Li, L. Mei, Chin. Chem. Lett. 31 (2020) 1345-1356. DOI:10.1016/j.cclet.2020.03.001 |
| [7] |
Q. Pan, C. Nie, Y. Hu, et al., ACS Appl. Mater. Interfaces 12 (2020) 400-409. DOI:10.1021/acsami.9b20707 |
| [8] |
J. Kim, C. Jo, W.G. Lim, et al., Adv. Mater. 30 (2018) e1707557. DOI:10.1002/adma.201707557 |
| [9] |
R. Fan, A. Tong, X. Li, et al., Int. J. Nanomedicine 10 (2015) 7291-7305. DOI:10.2147/IJN.S89066 |
| [10] |
J. Yang, S. Zhai, H. Qin, et al., Biomaterials 176 (2018) 1-12. DOI:10.1016/j.biomaterials.2018.05.033 |
| [11] |
Y. Chen, L. Li, W. Chen, et al., Chin. Chem. Lett. 30 (2019) 1353-1360. DOI:10.1016/j.cclet.2019.02.003 |
| [12] |
Y. Wang, Y. Song, G. Zhu, et al., Chin. Chem. Lett. 29 (2018) 1685-1688. DOI:10.1016/j.cclet.2017.12.004 |
| [13] |
D. Zhang, T. Wu, X. Qin, et al., Nano Lett. 19 (2019) 6635-6646. DOI:10.1021/acs.nanolett.9b02903 |
| [14] |
Z. Xiao, C. Ji, J. Shi, et al., Angew. Chem. Int. Ed. 51 (2012) 11853-11857. DOI:10.1002/anie.201204018 |
| [15] |
L. Qiu, T. Chen, I. Öçsoy, et al., Nano Lett. 15 (2015) 457-463. DOI:10.1021/nl503777s |
| [16] |
Y. Zhang, J. Chao, H. Liu, et al., Angew. Chem. Int. Ed. 55 (2016) 8036-8040. DOI:10.1002/anie.201512022 |
| [17] |
J. Cutler, E. Auyeung, C. Mirkin, J. Am. Chem. Soc. 134 (2012) 1376-1391. DOI:10.1021/ja209351u |
| [18] |
H. Yu, T. Man, W. Ji, et al., Chin. Chem. Lett. 30 (2019) 175-178. DOI:10.1016/j.cclet.2018.04.020 |
| [19] |
B. Liu, J. Liu, J. Am. Chem. Soc. 139 (2017) 9471-9474. DOI:10.1021/jacs.7b04885 |
| [20] |
A. Sosnik, R. Sodhi, P. Brodersen, et al., Biomaterials 27 (2006) 2340-2348. DOI:10.1016/j.biomaterials.2005.11.028 |
| [21] |
V. Bagalkot, O. Farokhzad, R. Langer, et al., Angew. Chem. Int. Ed. 45 (2006) 8149-8152. DOI:10.1002/anie.200602251 |
| [22] |
W. Sun, T. Jiang, Y. Lu, et al., J. Am. Chem. Soc. 136 (2014) 14722-14725. DOI:10.1021/ja5088024 |
| [23] |
H. Shi, T. Jin, J. Zhang, et al., Chin. Chem. Lett. 31 (2020) 155-158. DOI:10.1016/j.cclet.2019.06.020 |
| [24] |
S. Pal, S. Harmsen, A. Oseledchyk, et al., Adv. Funct. Mater. 27 (2017) 1606632. DOI:10.1002/adfm.201606632 |
| [25] |
G. Guo, S. Fu, L. Zhou, et al., Nanoscale 3 (2011) 3825-3832. DOI:10.1039/c1nr10484e |
| [26] |
X. Zhang, X. Fan, Y. Wang, et al., Anal. Chem. 92 (2020) 1455-1462. DOI:10.1021/acs.analchem.9b04660 |
| [27] |
L. Demers, C. Mirkin, R. Mucic, et al., Anal. Chem. 72 (2000) 5535-5541. DOI:10.1021/ac0006627 |
| [28] |
J. Choi, S. Kim, T. Tachikawa, et al., J. Am. Chem. Soc. 133 (2011) 16146-16153. DOI:10.1021/ja2061984 |
| [29] |
T. Chen, B. He, J. Tao, et al., Adv. Drug Deliv. Rev. 143 (2019) 177-205. DOI:10.1016/j.addr.2019.04.009 |
| [30] |
X. Zhang, C. Hu, C. Kong, et al., Cell Death Differ. 27 (2020) 540-555. DOI:10.1038/s41418-019-0372-z |
| [31] |
B. Wang, X. Yu, J. Wang, et al., Biomaterials 78 (2016) 27-39. DOI:10.1016/j.biomaterials.2015.11.025 |