 2021, Vol. 32
2021, Vol. 32 


b University of Science and Technology of China, Hefei 230026, China;
c Jilin Biomedical Polymers Engineering Laboratory, Changchun 130022, China;
d Department of Life Science and Technology, Tokyo Institute of Technology, Nagatsuta, Midori, Yokohama 226-8501, Japan
Programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) represented immune checkpoints blockade therapy, have achieved encouraging clinical results in different tumor types, including melanoma, bladder carcinoma and non-small cell lung [1-6]. Despite of these clinical achievements, many limitations should be further improved, such as the low objective response rate, which has been reported to be 10%–40% response rates generally, only the patients with immunogenic tumors could benefit from ICB therapy [7]. Moreover, patients often suffer from side effects including autoimmune diseases during ICB treatment for the antibodies non-specifically targeted to the normal cells [8-10]. Therefore, efforts to not only promote ICB response rate but also avoid side effects have become one of the crucial parts in the field of cancer immunotherapy.
To sensitize tumors to ICB therapy, recent reports have focused on the tumor vaccines [11-15], which could specifically activate the antitumor immune responses to enhance the immunogenicity of tumor cells [16-20]. The mechanism for tumor vaccines to boost immune system to fight against tumor cells could be explained. Tumor cells could release the antigens during their growth, and the antigens were then captured by dendritic cells (DCs), which could be further processed for antigens presentation. Next, the treated antigens were presented to T cells to prime the downstream antitumor T cell responses. The effector T cells then trafficked to and infiltrated into the tumors to specifically recognize and kill tumor cells [21]. Owing to the immune activation behavior of the tumor vaccines, many researches have combined tumor vaccines with ICB therapy for tumor suppression [22-24]. Although such combination strategy could improve the antitumor efficacy, the genetic mutations of tumor cells during tumor growth might lead to the antigen losses, which could limit the efficacy and applications of tumor vaccines [25, 26]. Besides, most the reported combination strategy of tumor vaccines and ICB therapy neglected the severe side effects of ICB therapy. Therefore, optimal strategy for combining tumor vaccines and ICB therapy to overcome these limitations should be proposed.
Herein, we developed an in situ vaccine approach by adjuvant unmethylated cytosine-phosphate-guanine (CpG) administration, which was a Toll-like receptor 9 (TLR9) ligand with potent immunostimulatory effects [27-29], and was delivered by polyethyleneimine (PEI) via simple electrostatic interactions. Such in situ vaccination strategy could avoid the problems for antigen losses during tumor growth. Furthermore, to combine anti-PD therapy for PD1/PD-L1 pathway blockade, we co-delivered the plasmid DNA expressing shRNA to downregulate PD-L1 gene with CpG by PEI delivery system, which was then shielded by aldehyde modified polyethylene glycol (OHC-PEG-CHO) via in situ pH responsive Schiff base reaction [30, 31]. The Schiff base bonds could specifically break under acidic tumor extracellular pH to rebound to higher positive potential to improve the cellular uptake of nanoparticles, the pH responsive delivery system we explored here could avoid the PD-L1 gene silencing in normal cells to reduce the side effects. The OHC-PEG-CHO/PEI/DNA/CpG nanoparticles (P(PDC) NPs) could be prepared within 30 min, such simple NPs achieved the combination treatment of in situ tumor vaccine and anti-PD therapy to realize superior antitumor efficacy in B16F10 tumor-bearing mice. In addition, the immune analysis revealed the amplified T cell responses enhancement and the innate immunity activation. The synergistic antitumor strategy we presented here provided a facile and effective combination treatment for tumor suppression (Scheme 1).

|
Download:
|
| Scheme 1. Schematic illustration of in situ vaccination and gene-mediated PD-L1 blockade for enhanced tumor immunotherapy. | |
{kind=link}
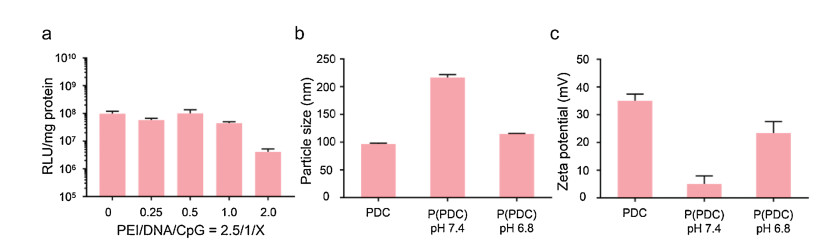
Firstly, in vitro transfection experiment was carried out to explore the optimum ratios of PEI/DNA/CpG. In comparison of the PEI/DNA complex, the little CpG addition caused the negligible decrease of the transfection efficacy in B16F10 cells, while the PDC NPs with higher CpG mass ratios exhibited obviously reduced transfection efficacy (Fig. 1a). Thus, the mass ratio of 2.5/1/0.5 for PEI/DNA/CpG was selected for the following experiments. In addition, the zeta potential of PDC NPs with different mass ratio was shown in Fig. S1 (Supporting information), more CpG led to lower zeta potential of PDC NPs, which suggested that the CpG could neutralize the positive charges of PEI/DNA complex to hinder the interactions between cells and nanoparticles.

|
Download:
|
| Fig. 1. (a) Transfection efficiency of various nanoparticles in B16F10 cells. (b) Particle size of different nanoparticles. (c) Zeta potential of different nanoparticles. | |
{kind=link}
We next examined the in situ shielding of OHC-PEG-CHO and the pH responsive behavior of the formed Schiff base bonds. The structure of the OHC-PEG-CHO was shown in Fig. S2 (Supporting information), which was synthesized as we described before and the 1H NMR of OHC-PEG-CHO indicated its successful synthesis [12]. The characterizations including particle size and zeta potential of nanoparticles after shielding were examined. The results showed that after OHC-PEG-CHO shielding, the prepared P(PDC) NPs at pH 7.4 exhibited larger particle size with 217 nm for DLS analysis (Fig. 1b), the elevated particle size might cause by PEG layers. To confirm the pH responsive behavior, the P(PDC) NPs was incubated in pH 6.8 buffer, then the particle size was decreased to 115 nm, which might attribute to the removal of the PEG layers under acidic environment. Furthermore, the zeta potential decreased after OHC-PEG-CHO shielding but rebounded in pH 6.8, confirming that the Schiff base bonds could break under acidic buffers for PEG detachment (Fig. 1c).
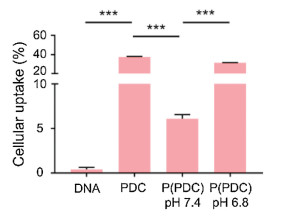
The cellular uptake behavior of nanoparticles was firstly performed by flow cytometry (Fig. 2). The results indicated that compared with the lowest cellular uptake of naked DNA, PDC exhibited much higher DNA fluorescence intensity, owing to the PEI delivery system for promoting uptake efficacy. P(PDC) in pH 7.4 showed the much lower DNA cellular uptake, which suggested that PEG shielding could prevent the interactions between nanoparticles and tumor cells for the decreased positive charges. P(PDC) in pH 6.8 caused obvious cellular uptake enhancement, confirming that the acidic environment could break the Schiff base bonds to detach the PEG shielding, thus exposing the positively charged PDC complexes to further interact with cells for internalization.

|
Download:
|
| Fig. 2. Cellular uptake of DNA, PDC, P(PDC) nanoparticles by FCM. | |
{kind=link}
The cellular internalization of nanoparticles was further examined by confocal laser scanning microscopy (CLSM). In accordance with the results for flow cytometry (FCM) analysis, reduced P(PDC) was found within tumor cells for pH 7.4 treatment, but remarkable elevated P(PDC) internalization was observed in pH 6.8 treated group (Fig. 3), confirming that acidic environment could remove the PEG shielding to promote nanoparticles internalization. The pH responsive delivery system might prevent the toxicity to the normal tissues for the specifically acidic tumor microenvironment.

|
Download:
|
| Fig. 3. CLSM images of tumor cells incubated with various formulations, DNA was labeled by Cy3, scale bar: 20 μm. | |
{kind=link}
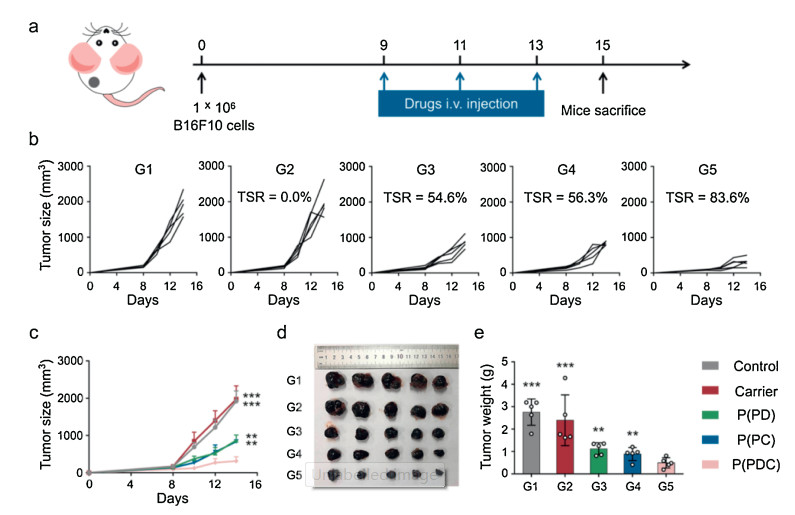
We next used mice-bearing B16F10 tumors, which was a highly aggressive tumor models, to validate the antitumor efficacy of the P(PDC)-based in situ vaccination and anti-PD combination therapy. All experimental procedures in this study were carried out on the basis of the guidelines established by Jilin University' Animal Care and Use Committee. Fig. 4a showed antitumor therapeutic schedule. At day 0, the B16F10 cells were subcutaneously injected to for B16F10 tumor models establishment. At day 9, when tumor sizes reached about 120 mm3, the tumor-bearing mice were sorted into five groups, then various formulations including PBS, Carrier (OHC-PEG-CHO/PEI), P(PD) (OHC-PEG-CHO/PEI/DNA), P(PC) (OHC-PEG-CHO/PEI/CpG), and P(PDC) NPs were respectively injected via tail vein every three day for total of three times. Until day 15, the mice were sacrificed for the tumor tissues collection, which would be examined for further immune analysis. The antitumor results were shown in Figs. 4b and c, PBS showed rapid tumor growth, while the PP treatments exhibited comparable tumor growth curves, indicating the negligible antitumor effects of the delivery system. P(PD) monotherapy caused the modest tumor suppression for the PD pathway blockade. Similarly, the P(PC) treatments induced moderate tumor inhibition, which might attribute to the activation of antitumor immunity after in situ vaccination. By contrast, the combined group achieved robust tumor inhibition, with 83.6% tumor suppression rate. Tumor images and tumor weights following various treatments also mirrored the antitumor curves (Figs. 4d and e). Moreover, hematoxylin-eosin (H & E) staining of tumor tissues at the end of antitumor treatments indicated that the combined group induced the most tumor cells necrosis (Fig. S3 in Supporting information). All of these results indicated that in situ vaccination could sensitize the tumors to anti-PD therapy to enhance the antitumor efficacy in highly aggressive B16F10 tumor models.

|
Download:
|
| Fig. 4. (a) In vivo antitumor experimental strategy. Individual (b) and average (c) tumor sizes in various groups. (d) Images of tumors after various treatments, TSR: tumor suppression rate. (e) Tumor weights during different antitumor treatments. G1, Control; G2, Carrier; G3, P(PD); G4, P(PC); G5, P(PDC). | |
{kind=link}
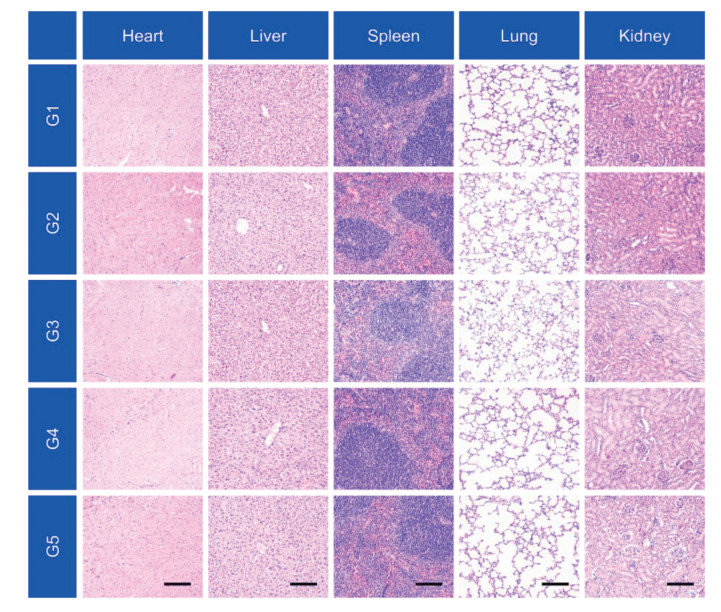
Moreover, the body weights during all antitumor treatments were not impacted, suggesting the superior biocompatibility of the combination strategy (Fig. S4 in Supporting information). To confirm these results, H & E staining of the major organs including heart, liver, spleen, lung and kidney were performed to analyze the histological damage following different treatments. Negligible histological damage was observed for treated groups, confirming the good biocompatibility of the designed treatments (Fig. 5). Moreover, side effects were not found in both P(PD) and P(PDC) group, for the tumor microenvironment responsive behavior, which was reported to avoid side effects to normal tissues [32]. These results revealed that the pH responsive delivery systems could achieve the PD-L1 silencing within tumor sites but avoid the severe side effects of the anti-PD therapy.

|
Download:
|
| Fig. 5. H & E staining of normal tissues after various treatments. Scale bar: 200 μm. G1, Control; G2, Carrier; G3, P(PD); G4, P(PC); G5, P(PDC). | |
{kind=link}
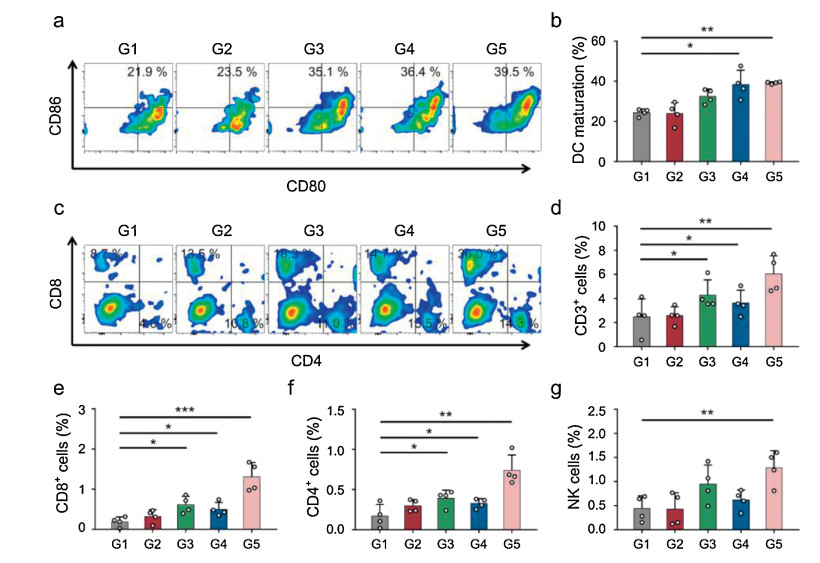
To explore the underlying immune mechanisms for the antitumor results, the intratumoral immune responses were detailedly analyzed following different treatments (Fig. S5 in Supporting information). As we all known, dendritic cells (DCs) played a crucial role for immune responses initiation and regulation [33, 34]. Thus, we firstly examined DCs activation within tumors for the in situ vaccination triggered by CpG administration. It was shown that tumors with CpG treatment exhibited obvious DCs maturation (CD11 c+ CD86+ CD80+) enhancement in comparison of control group, indicating that such in situ vaccination approach could effectively activated the DCs within tumors to initiate antitumor immunity (Figs. 6a and b). Meanwhile, we also measured the individual populations of T cells including cytotoxic T lymphocytes (CTLs, CD3+ CD8+ cells) and helper T cells (CD3+ CD4+ T cells), which were two main T lymphocytes for tumor killing [35-37]. Both P(PD) and P(PC) treatments caused moderate increase of CTLs and CD4 helper T cells infiltration. By contrast, the P(PDC) combination treatments induced the remarkable CTLs and CD4 helper T cells infiltration, even exhibiting 2.1- and 3.1-folds enhancement for CTLs infiltration in comparison of anti-PD and in situ vaccination monotherapy, respectively (Figs. 6c-f), in accompany with the increased levels of tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ) secretion after P(PDC) treatments (Fig. S6 in Supporting information), confirming that such two combined strategy could further amplified the antitumor T cell responses within tumors. Moreover, the natural killer cells (NK cells) within tumors showed the highest populations in combined group, which suggested that our designed in situ vaccination and anti-PD combined treatment could further activate innate antitumor immunity (Fig. 6g).

|
Download:
|
| Fig. 6. (a) Representative flow cytometric images of DCs maturation within tumors gating on CD11 c+ cells. (b) Proportions of DCs maturation gating on CD11 c + cells. (c) Representative quantitative analysis of T cells (gated on CD3+ T cells) by FCM detection. Proportions of (d) CD3+ T cells, (e) CTLs and (f) helper T cells within tumors. (g) Proportions of NK cells within tumors. G1, Control; G2, Carrier; G3, P(PD); G4, P(PC); G5, P(PDC). | |
{kind=link}
Since we have confirmed the activation of the antitumor immunity, we next examined weather such combination treatments could reverse the immunosuppressive tumor microenvironment (TME). We further evaluated the intratumoral populations of M2-like tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which have been reported to promote tumor growth [38, 39]. Slightly increase of M2-like TAMs and MDSCs was observed in the anti-PD monotherapy, but the CpG combination neutralized the increase and normalized the populations of MDSCs and M2-like TAMs, indicating that the combined strategy could not lead to the activation of the immunosuppressive TME (Figs. S7 and S8 in Supporting information). Moreover, the expression of PD-L1 has been obviously downregulated following combined treatments, which confirmed the PD pathway blockade (Fig. S9 in Supporting information).
In summary, we developed an in situ vaccination approach to promote the anti-PD therapy. PEI worked as delivery carrier to co-deliver the CpG and pshPD-L1, which was shielded by OHC-PEG-CHO to form the pH responsive P(PDC) NPs. CpG administration caused in situ tumor vaccine to promote the DCs activation, and activated the tumor immunity, which sensitized tumors to the anti-PD therapy. At final, CpG and PD-L1 blockade group exhibited the most tumor suppression behavior in B16F10 tumor-bearing mice. The immune analysis within tumors indicted the both amplified T cell responses and NK infiltration following combined treatments. Furthermore, the pH responsive P(PDC) NPs could prevent the side effects of the anti-PD therapy to the normal tissues for their lower cellular uptake efficacy but enhanced internalization efficacy owing to the removal of the PEG shielding under tumor acidic microenvironment. The synergistic antitumor strategy we presented might provide a facile and effective combination treatment for tumor inhibition.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors are thankful to the National Natural Science Foundation of China (Nos. 51925305, 51803210, 51520105004, 51873208, 51973217 and 51833010), Jilin province science and technology development program (Nos. 20200201075JC, 20180414027GH), National Science and Technology Major Projects for Major New Drugs Innovation and Development (No. 2018ZX09711003-012).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.055.
| [1] |
J.R. Brahmer, S.S. Tykodi, L.Q.M. Chow, et al., New Engl. J. Med. 366 (2012) 2455-2465. DOI:10.1056/NEJMoa1200694 |
| [2] |
M.E. Valsecchi, New Engl. J. Med. 373 (2015) 1270. DOI:10.1056/NEJMc1509660 |
| [3] |
J.M. Taube, A. Klein, J.R. Brahmer, et al., Clin. Cancer Res. 20 (2014) 5064-5074. DOI:10.1158/1078-0432.CCR-13-3271 |
| [4] |
K.M. Mahoney, P.D. Rennert, G.J. Freeman, Nat. Rev. Drug Discov. 14 (2015) 561-584. DOI:10.1038/nrd4591 |
| [5] |
M.E. Keir, M.J. Butte, G.J. Freeman, et al., Annu. Rev. Immunol. 26 (2008) 677-704. DOI:10.1146/annurev.immunol.26.021607.090331 |
| [6] |
P. Sharma, J.P. Allison, Science 348 (2015) 56-61. DOI:10.1126/science.aaa8172 |
| [7] |
R.S. Riley, C.H. June, R. Langer, et al., Nat. Rev. Drug Discov. 18 (2019) 175-196. DOI:10.1038/s41573-018-0006-z |
| [8] |
S.L. Topalian, J.M. Taube, R.A. Anders, et al., Nat. Rev. Cancer 16 (2016) 275-287. DOI:10.1038/nrc.2016.36 |
| [9] |
Q. Chen, C. Wang, G.J. Chen, et al., Adv. Healthc. Mater. 7 (2018) 1800424. DOI:10.1002/adhm.201800424 |
| [10] |
K.D. Moynihan, C.F. Opel, G.L. Szeto, et al., Nat. Med. 22 (2016) 1402-1410. DOI:10.1038/nm.4200 |
| [11] |
C. Wei, X. Dong, J. Liang, et al., J. Biomed. Nanotechnol. 15 (2019) 100-112. DOI:10.1166/jbn.2019.2669 |
| [12] |
Y.Y. Hu, L. Lin, J. Chen, et al., Biomaterials 252 (2020) 120114. DOI:10.1016/j.biomaterials.2020.120114 |
| [13] |
X.M. Yang, C.H. Lai, A.Q. Liu, et al., J. Biomed. Nanotechnol. 15 (2019) 1018-1032. DOI:10.1166/jbn.2019.2755 |
| [14] |
L. Zeng, Z. Liao, W. Li, et al., Chin. Chem. Lett. 31 (2020) 1162-1164. DOI:10.1016/j.cclet.2019.10.015 |
| [15] |
Z. Zhou, H. Lin, C. Li, et al., Chin. Chem. Lett. 29 (2018) 19-26. DOI:10.1016/j.cclet.2017.09.047 |
| [16] |
S.A. Rosenberg, J.C. Yang, N.P. Restifo, Nat. Med. 10 (2004) 909-915. DOI:10.1038/nm1100 |
| [17] |
J. Banchereau, A.K. Palucka, Nat. Rev. Immunol. 5 (2005) 296-306. DOI:10.1038/nri1592 |
| [18] |
P.A. Ott, Z.T. Hu, D.B. Keskin, et al., Nature 547 (2017) 217-221. DOI:10.1038/nature22991 |
| [19] |
D.J. Irvine, E.L. Dane, Nat. Rev. Immunol. 20 (2020) 321-334. DOI:10.1038/s41577-019-0269-6 |
| [20] |
L. Shen, T. Zhou, Y. Fan, et al., Chin. Chem. Lett. 31 (2020) 1709-1716. DOI:10.1016/j.cclet.2020.02.007 |
| [21] |
D.S. Chen, I. Mellman, Immunity 39 (2013) 1-10. DOI:10.1016/j.immuni.2013.07.012 |
| [22] |
J. Xu, H. Wang, L.G. Xu, et al., Biomaterials 207 (2019) 1-9. DOI:10.1016/j.biomaterials.2019.03.037 |
| [23] |
S.Y. Kim, S. Kim, J.E. Kim, et al., ACS Nano 13 (2019) 12671-12686. DOI:10.1021/acsnano.9b04207 |
| [24] |
X.Y. Ye, X. Liang, Q. Chen, et al., ACS Nano 13 (2019) 2956-2968. DOI:10.1021/acsnano.8b07371 |
| [25] |
M.M. Gubin, X.L. Zhang, H. Schuster, et al., Nature 515 (2014) 577-581. DOI:10.1038/nature13988 |
| [26] |
U. Sahin, E. Derhovanessian, M. Miller, et al., Nature 547 (2017) 222-226. DOI:10.1038/nature23003 |
| [27] |
C. Wang, W.J. Sun, G. Wright, et al., Adv. Mater. 28 (2016) 8912-8920. DOI:10.1002/adma.201506312 |
| [28] |
A. Marabelle, H. Kohrt, I. Sagiv-Barfi, et al., J. Clin. Invest. 123 (2013) 2447-2463. DOI:10.1172/JCI64859 |
| [29] |
Q. Xia, C. Gong, F. Gu, et al., J. Biomed. Nanotechnol. 14 (2018) 1613-1626. DOI:10.1166/jbn.2018.2605 |
| [30] |
X.W. Guan, Z.P. Guo, L. Lin, et al., Nano Lett. 16 (2016) 6823-6831. DOI:10.1021/acs.nanolett.6b02536 |
| [31] |
X.W. Guan, Z.P. Guo, T.H. Wang, et al., Biomacromolecules 18 (2017) 1342-1349. DOI:10.1021/acs.biomac.7b00080 |
| [32] |
W.M. Zhang, J. Zhang, Z. Qiao, et al., Chin. J. Polym. Sci. 36 (2018) 273-287. DOI:10.1007/s10118-018-2035-9 |
| [33] |
J. Banchereau, R.M. Steinman, Nature 392 (1998) 245-252. DOI:10.1038/32588 |
| [34] |
J. Banchereau, F. Briere, C. Caux, et al., Annu. Rev. Immunol. 18 (2000) 767-811. DOI:10.1146/annurev.immunol.18.1.767 |
| [35] |
R. Mahjub, S. Jatana, S.E. Lee, et al., J. Control. Release 288 (2018) 239-263. DOI:10.1016/j.jconrel.2018.09.010 |
| [36] |
S.R. Lu, N. Yang, J. He, et al., J. Biomed. Nanotechnol. 15 (2019) 593-601. DOI:10.1166/jbn.2019.2712 |
| [37] |
S. Ma, W.T. Song, Y.D. Xu, et al., Biomaterials 232 (2020) 119676. DOI:10.1016/j.biomaterials.2019.119676 |
| [38] |
L.L. Wu, H.L. Tang, H. Zheng, et al., J. Biomed. Nanotechnol. 15 (2019) 138-150. DOI:10.1166/jbn.2019.2661 |
| [39] |
D.I. Gabrilovich, S. Nagaraj, Nat. Rev. Immunol. 9 (2009) 162-174. DOI:10.1038/nri2506 |