 2021, Vol. 32
2021, Vol. 32 


b West China Hospital, Sichuan University, Chengdu 610041, China
Persistent infection caused by bacterial biofilms accounts for about 80% of nosocomial infections, which is a major hazard to human health [1-4]. Bacteria can readily colonize skin, teeth and internal organs to form biofilms, result of skin lesions, oral diseases and suppurative infections of organs [5-8]. Due to the shielding of extracellular polymeric substances (EPS), a hundredfold increase of antibiotic doses is required to eliminate bacterial biofilms [9-11]. More seriously, abuse of antibiotics has already induced the emergence of drug-resistant pathogenic bacteria, which severely threatens human's lives [12-16].
As well-known, amphiphilic quaternary ammonium salts (QASs) have been considered as effective antibacterial agents for both planktonic bacteria and biofilms [17-22]. The positive charge of QASs can readily target on the negative surface of bacterial cell membranes. Meanwhile, the hydrophobic alkyl chains of amphiphilic QASs can insert into the phospholipid bilayer, leading to the disruption of membrane structure and outflow of intracellular substances to cause death of bacteria [17, 19, 20]. The antibacterial mechanism of QASs is a kind of physical sterilization, which potentially inhibits the development of drug resistance [19, 23]. In addition, QAS-based antimicrobial nanoparticles (NPs) have been continuously reported to prevent and eliminate bacterial biofilms in vitro due to the penetration and decomposition of biofilms [22-26]. Despite the remarkable efficiency of combating planktonic bacteria and biofilms, the inherent toxicity of QASs, including cytotoxicity, protein absorption and hemolysis, severely limits the widespread application of disinfection in vivo [27-29]. Hence, most usage of QASs could only be found on design of antibacterial surfaces and dental materials [30-32]. In order to solve the toxic issues of QASs, many strategies have been proposed, such as variation of molecular structures [33-35], composited with biocompatible materials [5, 36] and shielding alkyl chains [17, 20] etc. However, some disadvantages are still existed, including tedious synthetic steps, high cost, low loading efficiency and QASs leaking. Recently, charge-convertible strategy (CCS) is gradually becoming prevalent in tumor diagnosis and therapy to promote long blood circulation and precise tumor targeting of drugs [19, 23, 37]. As the cytotoxicity of QASs is mainly derived from the positive moiety, we propose that controlled toxicity of QASs can be achieved by CCS to shield the positive charge and accomplish the bacterial disinfection application in vivo.
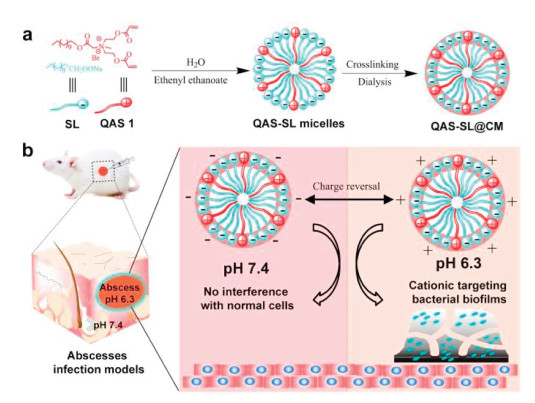
Herein, we developed a charge-convertible crosslinked micelle system by co-assembly of amphipathic QAS 1 and sodium laurate (SL), named QAS-SL@CM, for bacterial disinfection in vivo. As shown in Scheme 1, QAS 1 and SL can freely co-assemble to form negatively charged micelles to shield the positive charge. After cross-linking, QAS 1 could be firmly fixed in the micelles to restrict the movement of alkyl chains. Then the toxicity of the micelles system could be greatly reduced towards human cells [38]. By response of the acidic environment at infection sites, the surface charge of QAS-SL@CM could be immediately changed to positive due to the protonation of SL [17, 20]. Furthermore, β-thiopropionate bonds on QAS-SL@CM could also be disintegrated under acid environment to release QAS 1 and SL to kill bacteria both in vitro and in vivo [39, 40]. Therefore, through CCS and shell cross-linking strategy, a surface adaptive QAS-SL@CM could be constructed to intelligently respond to the microenvironment of bacterial infected wound site to solve the toxicity problem of QASs in vivo. The strategy reported here will provide new thoughts and methods for eliminating bacteria and biofilms, and has promising application of wound disinfection in clinic.

|
Download:
|
| Scheme1. (a) Schematic illustration for preparation of the quaternary ammonium salt and sodium laurate co-assembled crosslinked micelles (QAS-SL@CM) and (b) the treatment of subcutaneous abscesses infection in mice. | |
{kind=link}
Detailed synthesis and characterization of QAS 1 (Scheme S1 and Figs. S1–S3) can be found in Supporting information. In order to reduce the overall positive charge of QAS 1, a series of fatty acid sodium salts with various chain lengths (C10-C16) were selected to co-assemble with QAS 1 (Table S1, Figs. S4 and S5 in Supporting information). Dynamic light scattering (DLS) results showed that SL (C12) could co-assemble with QAS 1 to form stable micelles with particle size of ca. 59 nm and zeta potential of ca. −40 mV, named as QAS-SL. After cross-linked by dithiothreitol (DTT), co-assembled crosslinked micelles (QAS-SL@CM) could be formed with a diameter of ca. 52 nm (Fig. 1a and Table S1), which remained unchanged after incubation in water and serum for more than 24 h (Fig. S6 in Supporting information). Further tests showed that zeta potential of QAS-SL@CM could actually change from −25 mV to 30 mV under different pH conditions (5.0–8.2), while the particle size barely changed (Fig. 1b and Fig. S4). At pH 7.4, zeta potential showed −21 mV, as the negative charge of carboxylate in SL shielded the positive charge of QAS1 at the preset ratio. When pH was changed to 6.3, the overall charge of QAS-SL@CM was shifted to about 20 mV due to the protonation of carboxy group, indicating a charge-convertible process with pH variation. Transmission electron microscope (TEM) images showed that QAS@CM and QAS-SL@CM had uniform spherical shape with particle size of 38 and 45 nm (Figs. 1c and d), consistent with the data of DLS.

|
Download:
|
| Fig. 1. (a) Particle size of the samples (mean ± SD, n = 5). (b) The change of particle size and zeta potential under different pH conditions (mean ± SD, n = 5). (c, d) TEM images of QAS@CM and QAS-SL@CM. All samples were stained with an aqueous solution of 2% phosphotungstic acid. [QAS 1] = 2.5 mmol/L. | |
{kind=link}
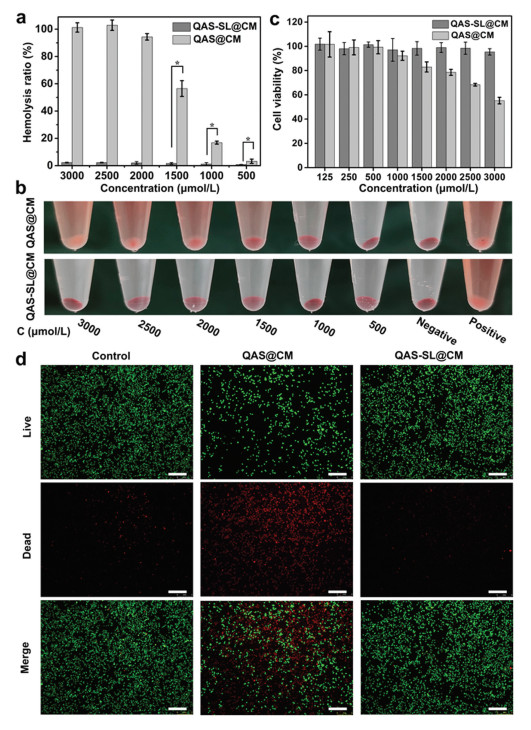
To test the biocompatibility of QAS-SL@CM, blood compatibility, cytotoxicity and protein adsorption capacity were characterized separately [38, 41]. As shown in Figs. 2a and b, QAS-SL@CM displayed good blood compatibility even at a high concentration of 3000 μmol/L with hemolysis rate less than 5%. In contrast, positively charged QAS@CM showed extremely higher hemolysis than QAS-SL@CM. Just at a low concentration of 500 μmol/L, the hemolysis rate was above 5%. At the same time, the solution almost completely turned red, indicating that a large number of cells had underwent hemolysis (Fig. 2b). Positive charge of QAS@CM can disturb the membrane structures of red cells to outflow inner contents to cause hemolysis. As the positive charges are shielded in QAS-SL@CM, the corresponding blood compatibility is greatly improved. Moreover, as shown in Fig. 2c, the cell compatibility of co-assembled QAS-SL@CM was also enhanced compared to that of QAS@CM. Meanwhile, the staining diagram of cells was represented in Fig. 2d after incubation with QAS-SL@CM and QAS@CM at a concentration of 3000 μmol/L. Due to the cytotoxicity of QAS@CM, about 50% dead cells could be found after incubation. While, almost no cell apoptosis was observed for QAS-SL@CM group just as the control group. Following, the adsorption capacity to BSA protein was also tested. As shown in Fig. S7 (Supporting information), a high protein adsorption rate of about 10% was obtained for positive QAS@CM after co-incubation for 6 h. For QAS-SL@CM, the adsorption rate did not exceed 5% even after increase the incubation time to 24 h. All the results above showed that the cytotoxicity, hemolytic toxicity and protein adsorption capacity could be controlled by co-assembly of QAS1 and SL.

|
Download:
|
| Fig. 2. Biocompatibility assessment assays of QAS-SL@CM and QAS@CM. (a) Optical photographs (mean ± SD, n = 5, *P < 0.05) and (b) Hemolysis ratio of blood cell test. (c) Cytotoxicity against L929 cells for 48 h incubation (mean ± SD, n = 5). (d) Fluorescence imaging of live/dead (green/red) staining of L929 cells incubated with QAS@CM and QAS-SL@CM. Scale bar: 200 μm. [QAS1] = 3000 μmol/L. | |
{kind=link}
In order to evaluate the biofilm elimination activity, S. aureus biofilm was firstly built, and then incubated with QAS@CM and QAS-SL@CM with various concentrations [1, 2, 42]. Crystal violet staining results in Figs. S8 a and b (Supporting information) showed that biofilm mass decreased with the increase of QAS1 concentration, corresponding to the colour gradually becoming lighter. As quantitatively characterized in Fig. S8 b, when the QAS 1 concentration increased to 110 μmol/L, almost all the biofilm was cleared and the optical absorbance approached zero. In addition, plate colony experiment in Fig. S8 c showed that almost no colonies existed after treated with QAS@CM and QAS-SL@CM at 110 μmol/L. Furthermore, the antibacterial mechanism of QAS-SL@CM was investigated by the live/dead staining of bacteria in biofilms. For QAS-SL@CM, there existed a balance between gain and loss of protons between lauric acid and SL in various acidic environments, resulting in different overall zeta potential (Fig. 3a). Numerous reports indicated that S. aureus were negatively charged, and the positively charged NPs were more likely to enter the interior of biofilm along pores in biofilms because of the charge attraction. As shown in Fig. 3b, buffer solutions of pH 7.4 and 6.3 were used to simulate the change of external environment to study the penetration of QAS-SL@CM and QAS@CM against S. aureus biofilms. At pH 7.4, QAS-SL@CM was mainly negative charged and not easy to enter the interior of biofilm. While, positive charged QAS-SL@CM at pH 6.3, was conducive to penetrate into the biofilm, leading to the death of bacteria (death rate 89%). For QAS@CM, the biofilm elimination efficiency was independent of pH, and the bacterial death rate of 85% and 83% could be obtained at pH 7.4 and 6.3 separately. The live bacteria were dyed green and the dead dyed red inside the biofilm. Also, scanning electron microscope (SEM) was conducted to observe the morphological change of S. aureus biofilms. As displayed in Fig. 3c, biofilms layers were destroyed severely and the number of bacteria greatly reduced for QAS-SL@CM and QAS@CM group, compared with the control group.

|
Download:
|
| Fig. 3. (a) Charge-convertible mechanism of QAS-SL@CM in different pH conditions. (b) Three-dimensional CLSM images of S. aureus biofilms incubated with different samples for 2 h. (c) SEM images of biofilms incubated with QAS@CM and QAS-SL@CM (100 μmol/L). | |
{kind=link}
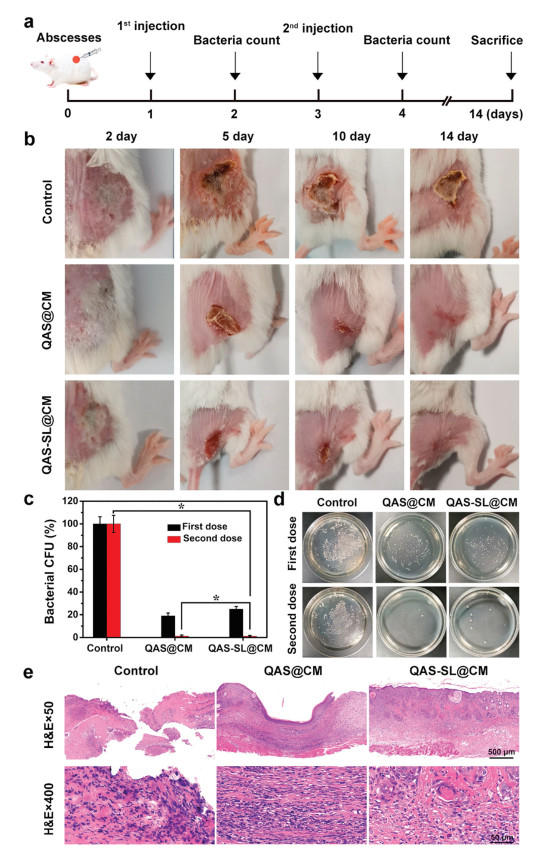
Furthermore, antibacterial application of QAS-SL@CM in vivo was studied in BALB/c mice with S. aureus infected subcutaneous abscess model (Approved by the Ethics Committee of Sichuan University) [43, 44]. Injection of saline and QAS@CM were applied as control groups, and the experimental design was shown in Fig. 4a. Images of abscesses sites were captured at various time after in-situ injection of saline, QAS@CM and QAS-SL@CM, separately. As displayed in Fig. 4b, serious suppuration could be found for saline treated group during the first five days, also accompanying with sharp weight loss (Fig. S9 in Supporting information). Even after 14 days, obvious skin damage could be observed due to the bacterial infection. For QAS@CM treated group, the abscesses volume decreased gradually with increase of time. However, still 10% of wound site could be observed after 14 days. While, the damaged skin for QAS-SL@CM treated group was almost completely healed for the same time. Meanwhile, no obvious weight loss could be found during the treatment period. In order to verify the bacterial elimination efficiency, pus in abscess site of mice was collected on day 2 and day 5. After plate colony incubation, bacterial colony was imaged and counted, which was shown in Figs. 4c and d. Large number of bacteria could be found on control group, even after 5 days' treatment. For QAS@CM and QAS-SL@CM group, bacteria were almost completely killed after 5 days, indicating good antibacterial effect, which agreed with the antibacterial results in vitro. All mice were sacrificed at 14th day and the skin tissues were collected for hematoxylin-eosin (H & E) staining. As shown in Fig. 4e, severe defects of skin epidermis could be observed for saline and QAS@CM groups. Meanwhile, there were large amounts of inflammatory cells dispersion. Though QAS@CM has good performance on bacterial elimination, the inherent cytotoxicity could trigger organism immune to induce inflammation response. In contrast, H & E slices of QAS-SL@CM treated group showed that dermal tissues at abscesses sites completely healed with less inflammatory cells. It was known that negatively charged QAS-SL@CM had good biocompatibility in vitro. Under acidic environment of bacterial infection, QAS 1 could be exposed to eliminate bacteria and biofilm due to charge-convertible. Thus, less inflammatory response could be induced during the treatment. Following, the pathological images were demonstrated by H & E assay of heart, liver, spleen, lung and kidney tissues, and shown in Fig. S10 (Supporting information). In contrast with normal mice, there were no apparent damage in major organs of QAS-SL@CM treated mice after 14 days.

|
Download:
|
| Fig. 4. (a) Establishment and treatment process of mouse subcutaneous abscesses models. (b) Optical photographs of abscesses in mice of different groups. (c) Histogram analysis and (d) bacterial plate tests of bacteria in abscesses of different groups (mean ± SD, n = 5, *P < 0.05). (e) Histological section analysis of subcutaneous abscesses models of different groups. | |
{kind=link}
In summary, charge-convertible quaternary ammonium salt-based micelles (QAS-SL@CM) were fabricated by the strategy of co-assembly of two amphiphiles with opposite charges. It is proved that the QAS-SL@CM presents lower cytotoxicity, hemolysis and protein adsorption compared with QAS@CM. Due to surface charge-convertible at acidic environment, QAS-SL@CM shows prominently antibacterial biofilms efficiency as QAS@CM. Significantly, experiments in vivo indicate that QAS-SL@CM not only has remarkable therapeutic effect of abscesses, but also performs no damages on major organs. This work may have guided significance in the development of QAS-based antibacterial materials for in vivo infection therapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21975165, 51703145 and 51673130), and China Postdoctoral Science Foundation (Nos. 2017M620426 and 2019T12084).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.034.
| [1] |
Y. Liu, H.J. Busscher, B. Zhao, et al., ACS Nano 10 (2016) 4779-4789. DOI:10.1021/acsnano.6b01370 |
| [2] |
Y. Wang, Y. Jin, W. Chen, et al., Chem. Eng. J. 358 (2019) 74-90. DOI:10.1016/j.cej.2018.10.002 |
| [3] |
J. Hoque, M.M. Konai, S.S. Sequeira, et al., J. Med. Chem. 59 (2016) 10750-10762. DOI:10.1021/acs.jmedchem.6b01435 |
| [4] |
L. Han, P. Li, P. Tang, et al., Nanoscale 11 (2019) 15846-15861. DOI:10.1039/C9NR03095F |
| [5] |
S. Goswami, D. Thiyagarajan, G. Das, et al., ACS Appl. Mater. Interfaces 6 (2014) 16384-16394. DOI:10.1021/am504779t |
| [6] |
Y. Yu, M. Cirelli, P. Li, et al., Ind. Eng. Chem. Res. 58 (2019) 21459-21465. DOI:10.1021/acs.iecr.9b03980 |
| [7] |
F.D. Victorelli, G.M.F. Calixto, M. Ramos, et al., J. Biomed. Nanotechnol. 14 (2018) 227-237. DOI:10.1166/jbn.2018.2484 |
| [8] |
L. Yan, T. Luan, Q. Hua, et al., J. Biomed. Nanotechnol. 14 (2018) 215-226. DOI:10.1166/jbn.2018.2500 |
| [9] |
K. Richter, P. Facal, N. Thomas, et al., ACS Appl. Mater. Interfaces 9 (2017) 21631-21638. DOI:10.1021/acsami.7b03672 |
| [10] |
Y. Li, K. Fukushima, D.J. Coady, et al., Angew. Chem. 52 (2013) 674-678. DOI:10.1002/anie.201206053 |
| [11] |
J.W. Costerton, P.S. Stewart, E.P. Greenberg, et al., Science 284 (1999) 1318-1322. DOI:10.1126/science.284.5418.1318 |
| [12] |
R.F. Landis, C.H. Li, A. Gupta, et al., J. Am. Chem. Soc. 140 (2018) 6176-6182. DOI:10.1021/jacs.8b03575 |
| [13] |
A. Harms, E. Maisonneuve, K. Gerdes, et al., Science 354 (2016) aaf4268. DOI:10.1126/science.aaf4268 |
| [14] |
H. Koo, R.N. Allan, R.P. Howlin, et al., Nat. Rev. Microbiol. 15 (2017) 740. DOI:10.1038/nrmicro.2017.99 |
| [15] |
Y. Yu, P. Li, C. Zhu, et al., Adv. Funct. Mater. 29 (2019) 1904402. DOI:10.1002/adfm.201904402 |
| [16] |
C. Liu, H. Shen, S. Wang, et al., Chin. Chem. Lett. 29 (2018) 1824-1828. DOI:10.1016/j.cclet.2018.10.025 |
| [17] |
F. Liu, D. He, Y. Yu, et al., Bioconjugate Chem. 30 (2019) 541-546. DOI:10.1021/acs.bioconjchem.9b00010 |
| [18] |
R. Joseph, A. Naugolny, M. Feldman, et al., J. Am. Chem. Soc. 138 (2016) 754-757. DOI:10.1021/jacs.5b11834 |
| [19] |
E. Obłąk, A. Piecuch, J. Rewak-Soroczynska, et al., Appl. Microbiol. Biotechnol. 103 (2019) 625-632. DOI:10.1007/s00253-018-9523-2 |
| [20] |
D. He, Y. Yu, F. Liu, et al., Chem. Eng. J. 382 (2020) 122976. DOI:10.1016/j.cej.2019.122976 |
| [21] |
P. Zhang, S. Li, H. Chen, et al., ACS Appl. Mater. Interfaces 9 (2017) 16933-16938. DOI:10.1021/acsami.7b05227 |
| [22] |
M.C. Jennings, L.E. Ator, T.J. Paniak, et al., ChemBioChem 15 (2014) 2211-2215. DOI:10.1002/cbic.201402254 |
| [23] |
S. Goswami, M.D. Adhikari, C. Kar, et al., J. Mater. Chem. 1 (2013) 2612-2623. DOI:10.1039/c3tb20226g |
| [24] |
L. Chu, H. Gao, T. Cheng, et al., Chem. Commun. 52 (2016) 6265-6268. DOI:10.1039/C6CC01269H |
| [25] |
C. Korupalli, C.C. Huang, W.C. Lin, et al., Biomaterials 116 (2017) 1-9. DOI:10.1016/j.biomaterials.2016.11.045 |
| [26] |
Y. Yao, D. Xu, Y. Zhu, et al., Chem. Sci. 11 (2020) 757-762. DOI:10.1039/C9SC05741B |
| [27] |
Y. Jiao, L.N. Niu, S. Ma, et al., Prog. Polym. Sci. 71 (2017) 53-90. DOI:10.1016/j.progpolymsci.2017.03.001 |
| [28] |
M. Nishida, S. Imazato, Y. Takahashi, et al., Biomaterials 31 (2010) 1518-1532. DOI:10.1016/j.biomaterials.2009.11.023 |
| [29] |
K. Kuroda, W.F. DeGrado, et al., J. Am. Chem. Soc. 127 (2005) 4128-4129. DOI:10.1021/ja044205+ |
| [30] |
Z. Li, X. Yang, H. Liu, et al., Chem. Eng. J. 374 (2019) 564-575. DOI:10.1016/j.cej.2019.05.208 |
| [31] |
S. Kumar Tiwari, X. Guo, Y. Huang, et al., Sci. Rep. 9 (2019) 12463. DOI:10.1038/s41598-019-48666-4 |
| [32] |
H.C. Pappas, S. Phan, S. Yoon, et al., ACS Appl. Mater. Interfaces 7 (2015) 27632-27638. DOI:10.1021/acsami.5b06852 |
| [33] |
A. Ivankin, L. Livne, A. Mor, et al., Angew. Chem. 49 (2010) 8462-8465. DOI:10.1002/anie.201003104 |
| [34] |
H. Sun, Y. Hong, Y. Xi, et al., Biomacromolecules 19 (2018) 1701-1720. DOI:10.1021/acs.biomac.8b00208 |
| [35] |
Y. Oda, S. Kanaoka, T. Sato, et al., Biomacromolecules 12 (2011) 3581-3591. DOI:10.1021/bm200780r |
| [36] |
Y. Zhang, X. He, M. Ding, et al., Biomacromolecules 19 (2018) 279-287. DOI:10.1021/acs.biomac.7b01016 |
| [37] |
Z. Yang, Y. Peng, L. Qiu, et al., Chin. Chem. Lett. 29 (2018) 1839-1844. DOI:10.1016/j.cclet.2018.11.009 |
| [38] |
T. Wang, G. Qu, Y. Deng, et al., Chin. Chem. Lett. 30 (2019) 2368-2374. DOI:10.1016/j.cclet.2019.11.021 |
| [39] |
Y. Yao, C. Li, F. Liu, et al., Phys. Chem. Chem. Phys. 21 (2019) 10477-10487. DOI:10.1039/C9CP01427F |
| [40] |
Y. Yao, Y. Chen, Y. Liu, et al., Langmuir 35 (2019) 5871-5877. DOI:10.1021/acs.langmuir.9b00607 |
| [41] |
P. Huang, J. Liu, W. Wang, et al., ACS Appl. Mater. Interfaces 6 (2014) 14631-14643. DOI:10.1021/am503974y |
| [42] |
J. Xia, W. Wang, X. Hai, et al., Chin. Chem. Lett. 30 (2019) 421-424. DOI:10.1016/j.cclet.2018.07.008 |
| [43] |
D. Hu, H. Li, B. Wang, et al., ACS Nano 11 (2017) 9330-9339. DOI:10.1021/acsnano.7b04731 |
| [44] |
H. Wang, W. Ouyang, X. Zhang, et al., J. Mater. Chem. B 7 (2019) 4630-4637. DOI:10.1039/C9TB00845D |